- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




The Open Virology Journal
(Discontinued)
ISSN: 1874-3579 ― Volume 15, 2021
Small Molecule Inhibitors of Human Papillomavirus Protein - Protein Interactions
C.M D’Abramo, J Archambault*
Abstract
Human papillomaviruses (HPV) have now been identified as a necessary cause of benign and malignant lesions of the differentiating epithelium, particularly cervical cancer, the second most prevalent cancer in women worldwide. While two prophylactic HPV vaccines and screening programs are available, there is currently no antiviral drug for the treatment of HPV infections and associated diseases. The recent progress toward the identification and characterization of specific molecular targets for small molecule-based approaches provides prospect for the development of effective HPV antiviral compounds. Traditionally, antiviral therapies target viral enzymes. HPV encode for few proteins, however, and rely extensively on the infected cell for completion of their life cycle. This article will review the functions of the viral E1 helicase, which encodes the only enzymatic function of the virus, of the E2 regulatory protein, and of the viral E6 and E7 oncogenes in viral replication and pathogenesis. Particular emphasis will be placed on the recent progress made towards the development of novel small molecule inhibitors that specifically target and inhibit the functions of these viral proteins, as well as their interactions with other viral and/or cellular proteins.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 5
First Page: 80
Last Page: 95
Publisher Id: TOVJ-5-80
DOI: 10.2174/1874357901105010080
Article History:
Received Date: 8/3/2011Revision Received Date: 23/5/2011
Acceptance Date: 13/6/2011
Electronic publication date: 4/7/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Laboratory of Molecular Virology, Institut de Recherches Cliniques de Montréal, 110 Pine Avenue West, Montreal, Quebec, H2W 1R7, Canada; Tel: (514) 987-5739; Fax: (514) 987-5741; E-mail: jacques.archambault@ircm.qc.ca
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 8-3-2011 |
Original Manuscript | Small Molecule Inhibitors of Human Papillomavirus Protein - Protein Interactions | |
HPV INFECTION AND ASSOCIATED DISEASES
Human papillomaviruses (HPV) are small, double-stranded DNA viruses that induce benign and malignant hyperproliferative lesions of the differentiating epithelium of the skin and mucosa. To date, more than 150 different HPV types have been identified [1de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses Virology 2004; 324: 17-27., 2Munger K, Baldwin A, Edwards KM, et al. Mechanisms of human papillomavirus-induced oncogenesis J Virol 2004; 78: 11451-60.], including approximately 30 to 40 types that preferentially infect the mucosa of the anogenital tract, causing a variety of abnormalities ranging from genital warts to invasive cancer. Of these, infection by oncogenic or high-risk HPV types such as HPV16,-18,-31,-33 or -45 is implicated in the development of cervical cancer [3Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues Nat Rev Cancer 2007; 7: 11-22.], one of the most common cancers among women worldwide, as well as other genital malignancies and a subset of head-and-neck cancers [4Gillison ML. Human papillomavirus-associated head and neck cancer is a distinct epidemiologic, clinical, and molecular entity Semin Oncol 2004; 31: 744-54., 5Syrjanen S. Human papillomavirus (HPV) in head and neck cancer J Clin Virol 2005; 32(Suppl 1): S59-66.]. Given that HPV DNA is detected in more than 99% of cancerous cervical lesions [6Walboomers JM, Jacobs MV, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide J Pathol 1999; 189: 12-9.], high-risk HPV strains are now well established as the causative agents for the development of cervical cancer [7Bosch FX, Lorincz A, Munoz N, Meijer CJ, Shah KV. The causal relation between human papillomavirus and cervical cancer J Clin Pathol 2002; 55: 244-65.] and many anal cancers [8Daling JR, Madeleine MM, Johnson LG, et al. Human papillomavirus, smoking, and sexual practices in the etiology of anal cancer Cancer 2004; 101: 270-80., 9Frisch M, Fenger C, van den Brule AJ, et al. Variants of squamous cell carcinoma of the anal canal and perianal skin and their relation to human papillomaviruses Cancer Res 1999; 59: 753-.]. HPV16 is the most prevalent, accounting for over 50% of cancerous lesions and their precursors [10Braaten KP, Laufer MR. Human Papillomavirus (HPV), HPV-related disease, and the HPV vaccine Rev Obstet Gynecol 2008; 1: 2-10., 11Clifford GM, Smith JS, Aguado T, Franceschi S. Comparison of HPV type distribution in high-grade cervical lesions and cervical cancer: a meta-analysis Br J Cancer 2003; 89: 101-5.]. Other low-risk HPV types such as HPV6 and -11 are also associated with specific pathological conditions including anogenital condyloma or benign genital warts [12Lacey CJ. Therapy for genital human papillomavirus-related disease J Clin Virol 2005; 32(Suppl 1): S82-90.], as well as recurrent laryngeal papillomatosis [13Gissmann L, Wolnik L, Ikenberg H, et al. Human papillomavirus types 6 and 11 DNA sequences in genital and laryngeal papillomas and in some cervical cancers Proc Natl Acad Sci USA 1983; 80: 560-3.]. More recently, studies have also suggested the involvement of cutaneous HPV types in a subset of non-melanoma skin cancers [14Akgul B, Cooke JC, Storey A. HPV-associated skin disease J Pathol 2006; 208: 165-75.].
Although HPV infection is quite common, the majority of these infections clear spontaneously with time. However, in some cases the infection will persist, a known risk factor for progression to malignancy. While carcinogenesis does not occur in the majority of infected individuals, cervical cancer is still the second leading cause of cancer-related deaths in women worldwide. There are an estimated 493,000 new cases of cervical cancer that result in 233,000 deaths per year worldwide [15Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics 2002 CA Cancer J Clin 2005; 55: 74-108.]. Annually, an estimated 11,070 women are diagnosed with invasive cervical cancer in the United States alone, and about 4000 women die from this disease [16Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008 CA Cancer J Clin 2008; 58: 71-96.]. Although not life-threatening, genital warts are also very common with approximately 1 million new cases diagnosed each year in the United States [10Braaten KP, Laufer MR. Human Papillomavirus (HPV), HPV-related disease, and the HPV vaccine Rev Obstet Gynecol 2008; 1: 2-10.]. The burden of HPV infections is greater in developing countries and accounts for approximately 83% of all new cases due to a lack of adequate screening programs [17Batson A, Meheus F, Brooke S. Chapter 26: Innovative financing mechanisms to accelerate the introduction of HPV vaccines in developing countries Vaccine 2006; 24(Suppl 3): S219-25.]. Despite the high incidence of HPV infection and its associated malignant diseases, there is currently no effective antiviral agent available for therapy. Current therapeutic approaches are usually aimed at the removal of HPV lesions through surgical excision or cryotherapy, making them mostly invasive and cytodestructive in nature [18Gross G. Therapy of human papillomavirus infection and associated epithelial tumors Intervirology 1997; 40: 368-77., 19Beutner KR, Ferenczy A. Therapeutic approaches to genital warts Am J Med 1997; 102: 28-37.]. A limited number of non-surgical treatments have been clinically approved including topical applications of imiquimod or podophyllin for treatment of genital warts [20Beutner KR, Tyring SK, Trofatter KF Jr, et al. Imiquimod, a patient-applied immune-response modifier for treatment of external genital warts AntimicrobAgents Chemother 1998; 42: 789-94., 21Scheinfeld N, Lehman DS. An evidence-based review of medical and surgical treatments of genital warts Dermatol Online J 2006; 12: 5.]. However, due to viral persistence and the limited efficacy of such non-specific treatments, rates of recurrence are high, especially for patients with immunologic deficiencies. The development of prophylactic HPV vaccines represents a major advance for the prevention of human cancers. In June 2006, the US FDA approved Gardasil® developed by Merck and Co. Inc. as the first HPV vaccine. This quadrivalent vaccine is based on recombinant virus-like particles (VLP) from HPV6,-11,-16, and -18 and has been shown to be highly effective in preventing pre-cancerous cervical lesions and genital warts caused by these HPV types [22Siddiqui MA, Perry CM. Human papillomavirus quadrivalent (types 6 11 16 18) recombinant vaccine (Gardasil) Drugs 2006; 66: 1263-71. discussion 72-3]. A second HPV vaccine, CervarixTM developed by GlaxoSmithKline has recently received FDA approval [23Monie A, Hung CF, Roden R, Wu TC. Cervarix: a vaccine for the prevention of HPV 16, 18-associated cervical cancer Biologics 2008; 2: 97-105.]. This vaccine is a bivalent VLP vaccine directed against the high-risk viruses HPV16 and -18 [10Braaten KP, Laufer MR. Human Papillomavirus (HPV), HPV-related disease, and the HPV vaccine Rev Obstet Gynecol 2008; 1: 2-10.]. Both vaccines seem to be effective in blocking infections against the most prevalent HPV types and are expected to significantly reduce persistent HPV infection and the incidence of cervical abnormalities in regions where vaccination rates are high [24Harper DM, Franco EL, Wheeler CM, et al. Sustained efficacy up to 4.5 years of a bivalent L1 virus-like particle vaccine against human papillomavirus types 16 and 18: follow-up from a randomised control trial Lancet 2006; 367: 1247-55.]. These vaccines have also been recently approved for use in men for the prevention of HPV-associated penile cancers and possibly for the prevention of oral cancers associated with high-risk HPV types [25Giuliano AR, Palefsky JM, Goldstone S, et al. Efficacy of quadrivalent HPV vaccine against HPV Infection and disease in males N Engl J Med 2011; 364: 401-11.]. They do not, however, offer protection against all HPV types, nor are they of therapeutic value in already infected patients. In fact, up to 30% of cervical cancers are caused by HPV types not covered by current vaccines [26Wheeler CM. HPV genotypes: implications for worldwide cervical cancer screening and vaccination Lancet Oncol 2010; 11: 1013-4.], emphasizing the need for the development of novel and effective therapy in combating the progression of HPV-associated diseases. This article reviews the recent progress made toward the development of such novel antiviral agents for the treatment of HPV infections, with emphasis on the identification of small molecule inhibitors that specifically target and inhibit essential HPV protein functions and viral-host protein interactions.
THE HPV LIFE CYCLE
Precise knowledge of the different stages of the viral life cycle of HPV has been well described and serves as a molecular basis for the development and evaluation of antiviral compounds for the treatment of HPV infections. Productive HPV infection is dependent upon the cellular differentiation program that keratinocytes undergo in the epithelium. HPV virions initially infect cells in the basal layer of the epithelium where their viral double-stranded DNA genome are maintained as autonomously replicating extrachromosomal elements (i.e., episomes) in the nucleus of infected cells (reviewed in [27Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity Rev Med Virol 2006; 16: 83-97., 28Doorbar J. The papillomavirus life cycle J Clin Virol 2005; 32(Suppl 1): S7-15.]). Following entry into basal epithelial cells, the virus takes advantage of the cellular replication machinery to allow for low levels of viral DNA synthesis, resulting in an episomal copy number of approximately 50-100 genomes per cell. Upon differentiation of infected cells, productive replication is established such that the viral genome is amplified to more than 1000 copies and expression of capsid proteins is induced, resulting in the synthesis of infectious virions that are assembled and released. The fact that efficient replication of the HPV episome relies on host cell factors that are expressed in S-phase, and thus not normally synthesized in differentiating post-mitotic keratinocytes, represents a particular challenge to the virus. To circumvent this problem, HPVs have developed a process mediated primarily by the viral gene products E6 and E7 to induce infected cells to enter S-phase while maintaining their differentiated state. The primary mean by which the viral oncogenes E6 and E7 exert this effect on the cell cycle of infected cells is by abrogating the p53 and retinoblastoma (Rb) tumor suppressor pathways, respectively. This cooperation between the two viral oncoproteins promotes a cellular environment supporting viral DNA replication by stimulating infected differentiating keratinocytes to re-enter S-phase (E7), while preventing p53-dependent cell cycle arrest and apoptotic response (E6) [27Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity Rev Med Virol 2006; 16: 83-97.].
Genomic organization is a well conserved feature among papillomaviruses. The HPV genome, approximately 8 Kb in size, contains three main regions, the early and late regions, based on the timing of viral protein expression following viral entry into the host cell, and the long control region (LCR). The early region encodes predominately for regulatory proteins that are essential for viral transcription and replication, as well as cell cycle control, contributing to the transformation and immortalization potential of HPV. The late region encodes for the two viral structural proteins, L1 and L2, needed for capsid formation. The LCR contains most of the regulatory DNA sequences needed for replication of the viral genome and for expression of the viral genes, including the origin of DNA replication and enhancer and promoter regions, respectively [27Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity Rev Med Virol 2006; 16: 83-97.]. In total, the HPV genome encodes eight well characterized proteins (Fig. 1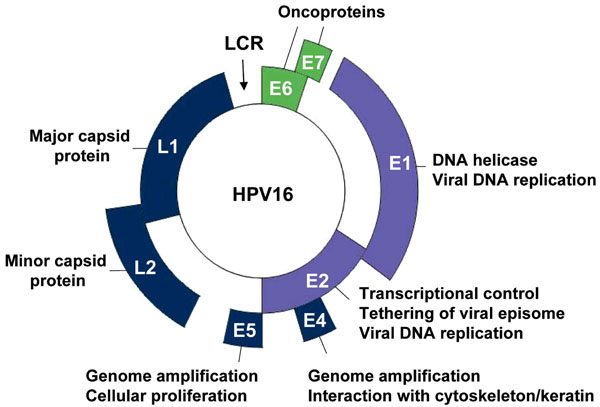 ).
).

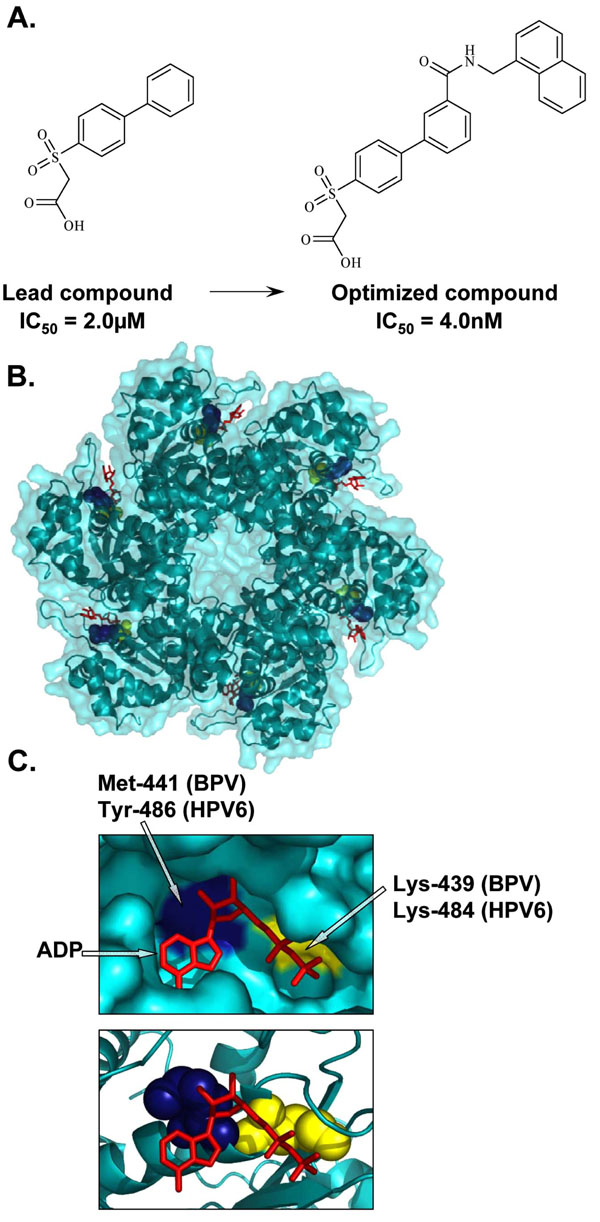 |
Fig. (3) Inhibition of E1 ATPase activity. (A) Structures of the lead and optimized biphenylsulfonacetic acid inhibitors of the E1 ATPase activity with IC50 values of 2.0µM and 4.0nM, respectively. (B) Crystal structure of the hexameric C-terminal helicase domain of bovine papillomavirus (BPV) E1 (PDB accession number 2GXA [173Enemark EJ, Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase Nature 2006; 442: 270-5.]). (C) Enlarged views of the ATP-binding pocket displaying the locations of the highly conserved catalytic Lys-439 (yellow), essential for ATP interaction and catalysis, and Met-441, (blue) important for the activity of biphenylsulfonacetic acid inhibitors. These residues are equivalent to Lys-484 and Tyr-486, respectively, in HPV6 E1. Bound ADP (red) is depicted in stick representation. |
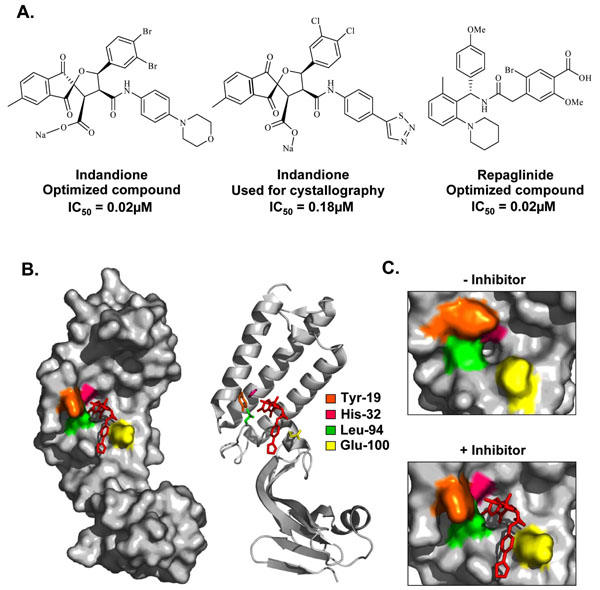 |
Fig. (4) Inhibition of the E1-E2 protein interaction. (A) Structures and potencies of optimized indandione and repaglinide inhibitors of the E1-E2 protein-protein interaction. (B) Surface and ribbon representation of the HPV11 E2 TAD-indandione inhibitor complex (PDB accession number 1R6K [101Wang Y, Coulombe R, Cameron DR, et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor J Biol Chem 2004; 279: 6976-85.]). The structure of the inhibitor used for crystallography is shown in (A). The amino acid residues that form the hydrophobic inhibitor-binding pocket are depicted in stick representation and are colored according to the legend in the figure. (C) Enlarged view of the hydrophobic pocket in the absence (left panel) and presence (right panel) of inhibitor, displaying the significant movement of the amino acids Tyr-19, His-32, Leu-94, and Glu-100 upon inhibitor binding. |
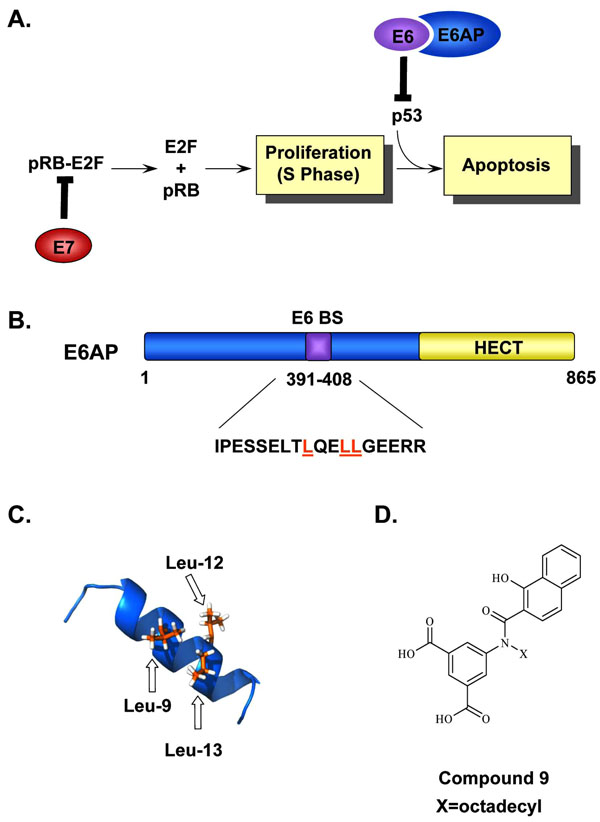 |
Fig. (5) Inhibition of the E6-E6AP interaction. (A) Simplified model of how the HPV oncogenes E6 and E7 stimulate cellular proliferation. Binding of E7 to pRb leads to the release and activation of the E2F transcription factors and drives differentiating keratinocytes into S-phase. This unscheduled DNA synthesis triggers a p53-dependent cell cycle arrest and apoptotic response that is prevented by E6, through its interaction with E6AP, and targets p53 for proteasomal degradation. (B) Schematic representation of E6AP. E6AP possesses several well-characterized functional domains including a HECT domain (yellow) and an E6-binding site (E6BS). The amino acid sequence corresponding to the E6BS is indicated and the three conserved leucine residues, Leu-9, Leu-12, Leu-13, are highlighted in orange. (C) NMR structure of the E6AP peptide showing the positions of the three leucine residues, Leu-9, Leu-12, and Leu-13 important for E6 binding are colored in orange (PDB accession number 1EQX [152Be X, Hong Y, Wei J, et al. Solution structure determination and mutational analysis of the papillomavirus E6 interacting peptide of E6AP Biochemistry 2001; 40: 1293-9.]). (D) Structure of Compound 9, an E6-E6AP inhibitor used as a starting point for the synthesis of other closely related inhibitors of E6 activity. |
E1 AND E2 PROTEINS AS ANTIVIRAL TARGETS
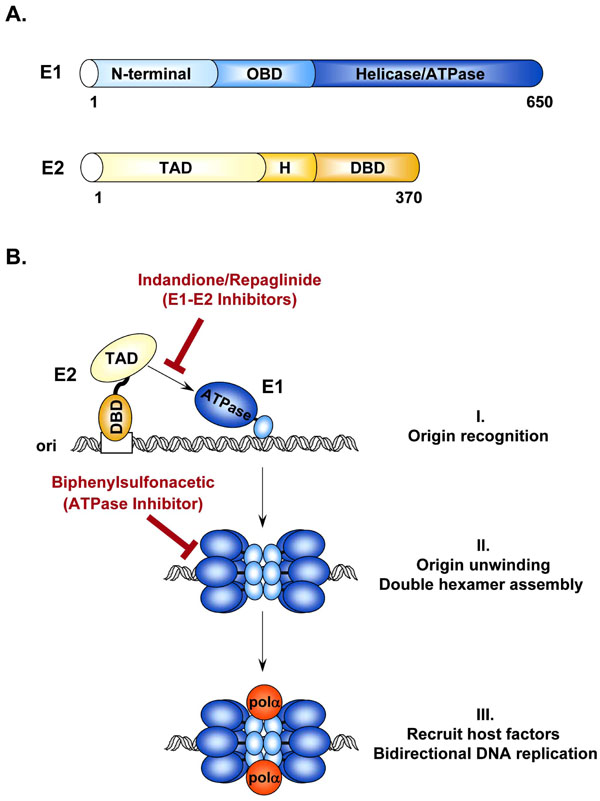
Viral replication and gene expression by the E1 and E2 proteins are obvious functions that could be targeted by small molecules for therapeutic intervention of HPV-associated diseases. Together with the cellular replication machinery, replication of the HPV genome requires the viral helicase E1 and the origin-binding protein E2. Structure-function studies have shown that E1 contains three functional domains: a C-terminal ATPase/helicase domain capable of oligomerization into hexamers [29Titolo S, Pelletier A, Pulichino AM, et al. Identification of domains of the human papillomavirus type 11 E1 helicase involved in oligomerization and binding to the viral origin J Virol 2000; 74: 7349-61., 30White PW, Pelletier A, Brault K, et al. Characterization of recombinant HPV6 and 11 E1 helicases: effect of ATP on the interaction of E1 with E2 and mapping of a minimal helicase domain J Biol Chem 2001; 276: 22426-38.], a central origin DNA-binding domain (OBD) [31Titolo S, Welchner E, White PW, Archambault J. Characterization of the DNA-binding properties of the origin-binding domain of simian virus 40 large T antigen by fluorescence anisotropy J Virol 2003; 77: 5512-8.], and an N-terminal regulatory region that is essential for replication in vivo but dispensable in vitro [32Amin AA, Titolo S, Pelletier A, et al. Identification of domains of the HPV11 E1 protein required for DNA replication in vitro Virology 2000; 272: 137-50.]. More specifically, this N-terminal region contains conserved sequences for nuclear localization (NLS), nuclear export (NES), a conserved cyclin-binding motif (CBM) that interacts with cyclin A/E-Cdk2 [33Ma T, Zou N, Lin BY, Chow LT, Harper JW. Interaction between cyclin-dependent kinases and human papillomavirus replication-initiation protein E1 is required for efficient viral replication Proc Natl Acad Sci USA 1999; 96: 382-7., 34Lin BY, Ma T, Liu JS, et al. HeLa cells are phenotypically limiting in cyclin E/CDK2 for efficient human papillomavirus DNA replication J Biol Chem 2000; 275: 6167-74.], as well as several phosphorylation sites for this kinase and others [33Ma T, Zou N, Lin BY, Chow LT, Harper JW. Interaction between cyclin-dependent kinases and human papillomavirus replication-initiation protein E1 is required for efficient viral replication Proc Natl Acad Sci USA 1999; 96: 382-7., 35Deng W, Lin BY, Jin G, et al. Cyclin/CDK regulates the nucleocytoplasmic localization of the human papillomavirus E1 DNA helicase J Virol 2004; 78: 13954-65., 36Fradet-Turcotte A, Moody C, Laimins LA, Archambault J. Nuclear export of human papillomavirus type 31 E1 is regulated by Cdk2 phosphorylation and required for viral genome maintenance J Virol 2010; 84: 11747-60.] (Fig. 2A ). As such, E1 functions both as a DNA binding protein to recognize the viral origin and as a helicase to unwind DNA ahead of the replication fork. Given its key role in HPV replication and the fact that it is the only enzymatic gene product encoded by the virus, E1 is undoubtedly an attractive target for the development of novel therapeutic agents. E2 is also considered a valid candidate target for antiviral compounds aimed at blocking viral DNA replication. E2 is a multifunctional protein that specifically binds to sites in the regulatory region of the viral genome to promote viral DNA replication, regulate viral gene transcription, and govern proper segregation of the viral episome to daughter cells at mitosis [37Hegde RS. The papillomavirus E2 proteins: structure, function, and biology Annu Rev Biophys Biomol Struct 2002; 31: 343-60.-41Howley PM, Lowy DR. Fields Virology. Philadelphia: Lippincott-Raven 2001; pp. 2197-29.]. The E2 protein is organized into two functional domains: an N-terminal transactivation domain (TAD) that is involved in transcriptional regulation and direct association with E1, and a C-terminal DNA-binding/dimerization domain (DBD). Both these domains are separated by a hinge region that is thought to be flexible and whose function has been poorly characterized (Fig. 2A). Recruitment of E1 to the origin is facilitated by its interaction with E2 [42Berg M, Stenlund A. Functional interactions between papillomavirus E1 and E2 proteins J Virol 1997; 71: 3853-63.-49Yang L, Li R, Mohr IJ, Clark R, Botchan MR. Activation of BPV-1 replication in vitro by the transcription factor E2 Nature 1991; 353: 628-32.], which binds to sites in the viral origin with high affinity (reviewed in [50McBride A, Myers G. The E2 proteins In: Myers G, Baker C, Mnger K, Sverdrup F, McBride A, Bernard H-U, Eds. In Human papillomavirus Theoretical Biology and Biophysics, Chapter III. New Mexico: Los Alamos 1997; pp. 53-73.]). Through these interactions, E2 not only facilitates recognition of the viral replication origin by E1 but also aids in the assembly of additional E1 proteins into replication-competent double hexamers necessary for bidirectional DNA unwinding. Through interactions with E1, cellular replication factors such as DNA polymerase α-primase [51Conger KL, Liu JS, Kuo SR, Chow LT, Wang TS. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human dna polymerase alpha/primase J Biol Chem 1999; 274: 2696-705.-53Park P, Copeland W, Yang L, et al. The cellular DNA polymerase alpha-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase Proc Natl Acad Sci USA 1994; 91: 8700-4.], topoisomerase I [54Clower RV, Fisk JC, Melendy T. Papillomavirus E1 protein binds to and stimulates human topoisomerase I J Virol 2006; 80: 1584-7.], and the single-stranded DNA binding protein RPA [55Loo YM, Melendy T. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA J Virol 2004; 78: 1605-5., 56Han Y, Loo YM, Militello KT, Melendy T. Interactions of the papovavirus DNA replication initiator proteins, bovine papillomavirus type 1 E1 and simian virus 40 large T antigen, with human replication protein A J Virol 1999; 73: 4899-907.] are recruited to the origin for assembly into an active replication complex (Fig. 2B). As such, both E1 and E2 are absolutely necessary for viral DNA replication in vivo [57Chiang CM, Ustav M, Stenlund A, et al. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins Proc Natl Acad Sci USA 1992; 89: 5799-803.]. Reverse genetic experiments have shown that both these viral proteins are essential for the maintenance of the viral episome in primary human keratinocyte cultures [45Frattini MG, Laimins LA. Binding of the human papillomavirus E1 origin-recognition protein is regulated through complex formation with the E2 enhancer-binding protein Proc Natl Acad Sci USA 1994; 91: 12398-402.] and for pathogenesis in the cottontail rabbit papillomavirus (CRPV) infection model [58Wu X, Xiao W, Brandsma JL. Papilloma formation by cottontail rabbit papillomavirus requires E1 and E2 regulatory genes in addition to E6 and E7 transforming genes J Virol 1994; 68: 6097-102.].
). As such, E1 functions both as a DNA binding protein to recognize the viral origin and as a helicase to unwind DNA ahead of the replication fork. Given its key role in HPV replication and the fact that it is the only enzymatic gene product encoded by the virus, E1 is undoubtedly an attractive target for the development of novel therapeutic agents. E2 is also considered a valid candidate target for antiviral compounds aimed at blocking viral DNA replication. E2 is a multifunctional protein that specifically binds to sites in the regulatory region of the viral genome to promote viral DNA replication, regulate viral gene transcription, and govern proper segregation of the viral episome to daughter cells at mitosis [37Hegde RS. The papillomavirus E2 proteins: structure, function, and biology Annu Rev Biophys Biomol Struct 2002; 31: 343-60.-41Howley PM, Lowy DR. Fields Virology. Philadelphia: Lippincott-Raven 2001; pp. 2197-29.]. The E2 protein is organized into two functional domains: an N-terminal transactivation domain (TAD) that is involved in transcriptional regulation and direct association with E1, and a C-terminal DNA-binding/dimerization domain (DBD). Both these domains are separated by a hinge region that is thought to be flexible and whose function has been poorly characterized (Fig. 2A). Recruitment of E1 to the origin is facilitated by its interaction with E2 [42Berg M, Stenlund A. Functional interactions between papillomavirus E1 and E2 proteins J Virol 1997; 71: 3853-63.-49Yang L, Li R, Mohr IJ, Clark R, Botchan MR. Activation of BPV-1 replication in vitro by the transcription factor E2 Nature 1991; 353: 628-32.], which binds to sites in the viral origin with high affinity (reviewed in [50McBride A, Myers G. The E2 proteins In: Myers G, Baker C, Mnger K, Sverdrup F, McBride A, Bernard H-U, Eds. In Human papillomavirus Theoretical Biology and Biophysics, Chapter III. New Mexico: Los Alamos 1997; pp. 53-73.]). Through these interactions, E2 not only facilitates recognition of the viral replication origin by E1 but also aids in the assembly of additional E1 proteins into replication-competent double hexamers necessary for bidirectional DNA unwinding. Through interactions with E1, cellular replication factors such as DNA polymerase α-primase [51Conger KL, Liu JS, Kuo SR, Chow LT, Wang TS. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human dna polymerase alpha/primase J Biol Chem 1999; 274: 2696-705.-53Park P, Copeland W, Yang L, et al. The cellular DNA polymerase alpha-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase Proc Natl Acad Sci USA 1994; 91: 8700-4.], topoisomerase I [54Clower RV, Fisk JC, Melendy T. Papillomavirus E1 protein binds to and stimulates human topoisomerase I J Virol 2006; 80: 1584-7.], and the single-stranded DNA binding protein RPA [55Loo YM, Melendy T. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA J Virol 2004; 78: 1605-5., 56Han Y, Loo YM, Militello KT, Melendy T. Interactions of the papovavirus DNA replication initiator proteins, bovine papillomavirus type 1 E1 and simian virus 40 large T antigen, with human replication protein A J Virol 1999; 73: 4899-907.] are recruited to the origin for assembly into an active replication complex (Fig. 2B). As such, both E1 and E2 are absolutely necessary for viral DNA replication in vivo [57Chiang CM, Ustav M, Stenlund A, et al. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins Proc Natl Acad Sci USA 1992; 89: 5799-803.]. Reverse genetic experiments have shown that both these viral proteins are essential for the maintenance of the viral episome in primary human keratinocyte cultures [45Frattini MG, Laimins LA. Binding of the human papillomavirus E1 origin-recognition protein is regulated through complex formation with the E2 enhancer-binding protein Proc Natl Acad Sci USA 1994; 91: 12398-402.] and for pathogenesis in the cottontail rabbit papillomavirus (CRPV) infection model [58Wu X, Xiao W, Brandsma JL. Papilloma formation by cottontail rabbit papillomavirus requires E1 and E2 regulatory genes in addition to E6 and E7 transforming genes J Virol 1994; 68: 6097-102.].
In addition to its role in replication, E2 is also implicated in the regulation of viral gene transcription and segregation of the episome at mitosis [37Hegde RS. The papillomavirus E2 proteins: structure, function, and biology Annu Rev Biophys Biomol Struct 2002; 31: 343-60., 39Ilves I, Kivi S, Ustav M. Long-term episomal maintenance of bovine papillomavirus type 1 plasmids is determined by attachment to host chromosomes, which Is mediated by the viral E2 protein and its binding sites J Virol 1999; 73: 4404-12.]. Depending on the promoter context, E2 has either activating or repressing functions. For instance, E2 activates transcription from a minimal promoter under the control of multimerized E2-binding sites [59Kovelman R, Bilter GK, Glezer E, Tsou AY, Barbosa MS. Enhanced transcriptional activation by E2 proteins from the oncogenic human papillomaviruses J Virol 1996; 70: 7549-60.], while in the context of the viral genome, E2 represses viral transcription driven from the LCR during the early stages of viral infection, particularly of the E6 and E7 genes [59Kovelman R, Bilter GK, Glezer E, Tsou AY, Barbosa MS. Enhanced transcriptional activation by E2 proteins from the oncogenic human papillomaviruses J Virol 1996; 70: 7549-60.-63Soeda E, Ferran MC, Baker CC, McBride AA. Repression of HPV16 early region transcription by the E2 protein Virology 2006; 351: 29-41.]. Given its role as a transcriptional regulator, E2 has been shown to associate with a number of cellular transcription factors including TFIIB [64Rank NM, Lambert PF. Bovine papillomavirus type 1 E2 transcriptional regulators directly bind two cellular transcription factors, TFIID and TFIIB J Virol 1995; 69: 6323-34.-66Yao JM, Breiding DE, Androphy EJ. Functional interaction of the bovine papillomavirus E2 transactivation domain with TFIIB J Virol 1998; 72: 1013-9.], TBP and TFIID [64Rank NM, Lambert PF. Bovine papillomavirus type 1 E2 transcriptional regulators directly bind two cellular transcription factors, TFIID and TFIIB J Virol 1995; 69: 6323-34., 67Steger G, Ham J, Lefebvre O, Yaniv M. The bovine papillomavirus 1 E2 protein contains two activation domains: one that interacts with TBP and another that functions after TBP binding EMBO J 1995; 14: 329-40.-69Carrillo E, Garrido E, Gariglio P. Specific in vitro interaction between papillomavirus E2 proteins and TBP-associated factors Intervirology 2004; 47: 342-9.], AMF-1/GPS2 [70Breiding DE, Sverdrup F, Grossel MJ, et al. Functional interaction of a novel cellular protein with the papillomavirus E2 transactivation domain Mol Cell Biol 1997; 17: 7208-19.], p300/CBP [71Lee D, Lee B, Kim J, Kim DW, Choe J. cAMP response element-binding protein-binding protein binds to human papillomavirus E2 protein and activates E2-dependent transcription J Biol Chem 2000; 275: 7045-51., 72Muller A, Ritzkowsky A, Steger G. Cooperative activation of human papillomavirus type 8 gene expression by the E2 protein and the cellular coactivator p300 J Virol 2002; 76: 11042-53.], NAP-1 [73Rehtanz M, Schmidt HM, Warthorst U, Steger G. Direct interaction between nucleosome assembly protein 1 and the papillomavirus E2 proteins involved in activation of transcription Mol Cell Biol 2004; 24: 2153-68.], P/CAF [74Lee D, Hwang SG, Kim J, Choe J. Functional interaction between p/CAF and human papillomavirus E2 protein J Biol Chem 2002; 277: 6483-9.], and TopBP1 [75Boner W, Taylor ER, Tsirimonaki E, et al. A Functional interaction between the human papillomavirus 16 transcription/replication factor E2 and the DNA damage response protein TopBP1 J Biol Chem 2002; 277: 22297-303.]. More recent studies have demonstrated that the bromodomain-containing protein 4 (Brd4), a chromatin associated factor, is crucial for both the transactivation and repression functions of E2 [76Schweiger MR, You J, Howley PM. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function J Virol 2006; 80: 4276-85.-78Wu SY, Lee AY, Hou SY, et al. Brd4 links chromatin targeting to HPV transcriptional silencing Genes Dev 2006; 20: 2383-96.]. As a segregation factor, E2 tethers the viral episome to mitotic chromatin [39Ilves I, Kivi S, Ustav M. Long-term episomal maintenance of bovine papillomavirus type 1 plasmids is determined by attachment to host chromosomes, which Is mediated by the viral E2 protein and its binding sites J Virol 1999; 73: 4404-12., 79Lehman CW, Botchan MR. Segregation of viral plasmids depends on tethering to chromosomes and is regulated by phosphorylation Proc Natl Acad Sci USA 1998; 95: 4338-.-81Bastien N, McBride AA. Interaction of the papillomavirus E2 protein with mitotic chromosomes Virology 2000; 270: 124-34.] and possibly also to the mitotic spindle [82Dao LD, Duffy A, Van Tine BA, et al. Dynamic localization of the human papillomavirus type 11 origin binding protein E2 through mitosis while in association with the spindle apparatus J Virol 2006; 80: 4792-800., 83Van Tine BA, Dao LD, Wu SY, et al. Human papillomavirus (HPV) origin-binding protein associates with mitotic spindles to enable viral DNA partitioning Proc Natl Acad Sci USA 2004; 101: 4030-5.]. Biochemical assays have identified Brd4 as the E2 interacting protein responsible for chromosome tethering [40McBride AA, McPhillips MG, Oliveira JG. Brd4: tethering, segregation and beyond Trends Microbiol 2004; 12: 527-9., 76Schweiger MR, You J, Howley PM. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function J Virol 2006; 80: 4276-85., 78Wu SY, Lee AY, Hou SY, et al. Brd4 links chromatin targeting to HPV transcriptional silencing Genes Dev 2006; 20: 2383-96., 84You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes Cell 2004; 117: 349-60.-87Brannon AR, Maresca JA, Boeke JD, Basrai MA, McBride AA. Reconstitution of papillomavirus E2-mediated plasmid maintenance in Saccharomyces cerevisiae by the Brd4 bromodomain protein Proc Natl Acad Sci USA 2005; 102: 2998-3003.]. The possibility that all papillomavirus types rely on Brd4 for segregation or that alternative mechanisms exist is currently a subject of intense studies [88McPhillips MG, Ozato K, McBride AA. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin J Virol 2005; 79: 8920-32.-90Baxter MK, McPhillips MG, Ozato K, McBride AA. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4 J Virol 2005; 79: 4806-18.]. Mutational analyses of the E2 TAD have revealed that substitutions at amino acids arginine 37 and isoleucine 73 specifically abrogate binding to Brd4. These mutations inhibit the transcriptional activity of E2 but have little effect on its ability to support replication and maintenance of the viral episome in HPV immortalized keratinocyte cultures, and to complete the viral life cycle in organotypic raft cultures [76Schweiger MR, You J, Howley PM. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function J Virol 2006; 80: 4276-85., 77Senechal H, Poirier GG, Coulombe B, Laimins LA, Archambault J. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4 Virology 2007; 358: 10-7., 86Ilves I, Maemets K, Silla T, Janikson K, Ustav M. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle J Virol 2006; 80: 3660-5.]. In contrast, similar mutations in CRPV E2 were found to completely abolish the ability of the cloned CRPV genome to induce epithelial tumors in rabbits [91Jeckel S, Huber E, Stubenrauch F, Iftner T. A transactivator function of cottontail rabbit papillomavirus e2 is essential for tumor induction in rabbits J Virol 2002; 76: 11209-5.], suggesting an essential role for the interaction of E2 with Brd4 in viral pathogenesis.
Overall, it is clear that both the E1 and E2 proteins represent valid targets for therapeutic intervention. Given that these proteins are relatively well conserved in sequence and in function suggests that the use of small molecule-based approaches to interfere with the helicase/ATPase activity of E1, the E1-E2 interaction, or even the binding of E2 to Brd4 should be effective for the treatment of infections caused by different HPV types.
E1 ATPASE INHIBITORS
HPV lack the enzymes generally targeted by currently available antiviral agents, such as proteases and polymerases. E1 is the only enzymatic gene product with ATPase and DNA helicase activities that facilitate unwinding of the viral DNA ahead of the replication fork. Inhibition of this unwinding activity, powered mainly by the hydrolysis of ATP [92Rocque WJ, Porter DJ, Barnes JA, et al. Replication-associated activities of purified human papillomavirus type 11 E1 helicase Protein Expr Purif 2000; 18: 148-59.], would essentially block viral replication. Furthermore, since ATP promotes the oligomerization of E1 into hexamers, which in turn affects the interaction of E1 with E2, ATP can be viewed as an allosteric modulator of the E1-E2 protein interaction. The ATPase and unwinding activities are located within the C-terminal region of E1, the same region that binds to E2. Studies have shown that E2 binding hinders the ATPase activity of E1 and similarly, that ATP weakens the E1-E2 interaction and impairs the cooperative binding of E1 and E2 to the origin [30White PW, Pelletier A, Brault K, et al. Characterization of recombinant HPV6 and 11 E1 helicases: effect of ATP on the interaction of E1 with E2 and mapping of a minimal helicase domain J Biol Chem 2001; 276: 22426-38.]. These findings led to the proposal that ATP, in addition to powering the helicase activity of E1, also acts as a molecular trigger during the initiation of viral DNA replication to help weaken the E1-E2 interaction and promote E1 hexamerization at the origin [30White PW, Pelletier A, Brault K, et al. Characterization of recombinant HPV6 and 11 E1 helicases: effect of ATP on the interaction of E1 with E2 and mapping of a minimal helicase domain J Biol Chem 2001; 276: 22426-38., 47Sanders CM, Stenlund A. Recruitment and loading of the E1 initiator protein: an ATP-dependent process catalysed by a transcription factor EMBO J 1998; 17: 7044-55.]. As such, interfering with ATP binding and/or ATPase activity would result in the inhibition of HPV DNA replication and would be a valid strategy for the development of HPV antivirals. Small molecule inhibitors targeting the ATPase activity of HPV6 E1 have been identified by high-throughput screening of a large compound collection of greater than 500,000 diverse chemical entities [93Faucher AM, White PW, Brochu C, et al. Discovery of small-molecule inhibitors of the ATPase activity of human papillomavirus E1 helicase J Med Chem 2004; 47: 18-21.]. Lead molecules consisted of biphenylsulfonacetic acid analogues characterized by the presence of a biphenyl group substituted with a sulfonylacetic acid moiety [93Faucher AM, White PW, Brochu C, et al. Discovery of small-molecule inhibitors of the ATPase activity of human papillomavirus E1 helicase J Med Chem 2004; 47: 18-21.] (Fig. 3A ). Kinetic studies revealed that the mode of action of these compounds is characteristic of a hyperbolic competitive mechanism, suggesting that they do not bind directly at the ATP-binding site, but affect ATP binding through an allosteric mechanism [94White PW, Faucher AM, Massariol MJ, et al. Biphenylsulfonacetic acid inhibitors of the human papillomavirus type 6 E1 helicase inhibit ATP hydrolysis by an allosteric mechanism involving tyrosine 486 Antimicrob Agents Chemother 2005; 49: 4834-2.]. Thus, the activity of these inhibitors is a function of the ATP concentration. Given that the biphenyl and sulfonylacetic acid moieties were determined to be important for potency, both pharmacophores were subjected to chemical modifications for the synthesis of derivatives with improved potency, with IC50 values as low as 4 nM (Fig. 3A). Of interest was the finding that the addition of substituted amides at the 3’ and 4’ positions of the biphenyl moiety significantly improved the affinity of the original lead inhibitor for HPV6 E1 [93Faucher AM, White PW, Brochu C, et al. Discovery of small-molecule inhibitors of the ATPase activity of human papillomavirus E1 helicase J Med Chem 2004; 47: 18-21.]. These modifications, however, only slightly improved the affinity of these compounds for the related HPV11 E1 protein. In fact, they were 2- to 10-fold less active against the ATPase activity of HPV11 E1 compared to that of HPV6 E1. Given the high level of conservation between the helicase domains of these two proteins, a series of mutagenesis studies were conducted to identify amino acids within E1 important for inhibition [94White PW, Faucher AM, Massariol MJ, et al. Biphenylsulfonacetic acid inhibitors of the human papillomavirus type 6 E1 helicase inhibit ATP hydrolysis by an allosteric mechanism involving tyrosine 486 Antimicrob Agents Chemother 2005; 49: 4834-2.]. This led to the identification of Tyr-486, which is a cysteine in HPV11, as the single critical residue in HPV6 E1 responsible for inhibitor potency and binding. This was further substantiated by the finding that substitution of Cys-486 for tyrosine in HPV11 E1 substantially increased its sensitivity to the inhibitors. Interestingly, HPV18 E1 also contains a tyrosine at this position and accordingly could be inhibited by this class of inhibitors, although at a reduced potency compared to HPV6 E1. This latter finding raises the possibility that this class of small molecule inhibitors could be optimized for activity against many HPV types [94White PW, Faucher AM, Massariol MJ, et al. Biphenylsulfonacetic acid inhibitors of the human papillomavirus type 6 E1 helicase inhibit ATP hydrolysis by an allosteric mechanism involving tyrosine 486 Antimicrob Agents Chemother 2005; 49: 4834-2.]. Using the crystal structure of the HPV18 E1 helicase domain as a tool to determine the possible binding region of these inhibitors revealed that Tyr-486 in HPV6 E1 is located close to the highly conserved Lys-484, an essential residue for ATP interaction and catalysis [95Abbate EA, Berger JM, Botchan MR. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2 Genes Dev 2004; 18: 1981-96.] (Fig. 3B, C). It has therefore been speculated that the interaction of these inhibitors with Tyr-486 may induce a conformational change in E1 that would indirectly affect ATP binding, accounting for their allosteric mode of action. Consistent with the fact that ATP hydrolysis is required for helicase activity, the inhibitors also inhibited the helicase activity of HPV6 E1 in vitro. Despite these promising results, the biphenyl-sulfonacetic acid inhibitors were not active in cell-based assays [93Faucher AM, White PW, Brochu C, et al. Discovery of small-molecule inhibitors of the ATPase activity of human papillomavirus E1 helicase J Med Chem 2004; 47: 18-21.], perhaps attributable in part to the relatively high intracellular concentration of ATP. Nonetheless, this class of inhibitors has demonstrated the potential of the E1 ATPase domain as a target for inhibition of HPV DNA replication by small molecules. The identification of the small molecule binding pocket on the HPV E1 protein by X-ray crystallo-graphy of an E1-inhibitor complex would be the next logical step towards the design of second generation inhibitors with increased specificity and activity.
). Kinetic studies revealed that the mode of action of these compounds is characteristic of a hyperbolic competitive mechanism, suggesting that they do not bind directly at the ATP-binding site, but affect ATP binding through an allosteric mechanism [94White PW, Faucher AM, Massariol MJ, et al. Biphenylsulfonacetic acid inhibitors of the human papillomavirus type 6 E1 helicase inhibit ATP hydrolysis by an allosteric mechanism involving tyrosine 486 Antimicrob Agents Chemother 2005; 49: 4834-2.]. Thus, the activity of these inhibitors is a function of the ATP concentration. Given that the biphenyl and sulfonylacetic acid moieties were determined to be important for potency, both pharmacophores were subjected to chemical modifications for the synthesis of derivatives with improved potency, with IC50 values as low as 4 nM (Fig. 3A). Of interest was the finding that the addition of substituted amides at the 3’ and 4’ positions of the biphenyl moiety significantly improved the affinity of the original lead inhibitor for HPV6 E1 [93Faucher AM, White PW, Brochu C, et al. Discovery of small-molecule inhibitors of the ATPase activity of human papillomavirus E1 helicase J Med Chem 2004; 47: 18-21.]. These modifications, however, only slightly improved the affinity of these compounds for the related HPV11 E1 protein. In fact, they were 2- to 10-fold less active against the ATPase activity of HPV11 E1 compared to that of HPV6 E1. Given the high level of conservation between the helicase domains of these two proteins, a series of mutagenesis studies were conducted to identify amino acids within E1 important for inhibition [94White PW, Faucher AM, Massariol MJ, et al. Biphenylsulfonacetic acid inhibitors of the human papillomavirus type 6 E1 helicase inhibit ATP hydrolysis by an allosteric mechanism involving tyrosine 486 Antimicrob Agents Chemother 2005; 49: 4834-2.]. This led to the identification of Tyr-486, which is a cysteine in HPV11, as the single critical residue in HPV6 E1 responsible for inhibitor potency and binding. This was further substantiated by the finding that substitution of Cys-486 for tyrosine in HPV11 E1 substantially increased its sensitivity to the inhibitors. Interestingly, HPV18 E1 also contains a tyrosine at this position and accordingly could be inhibited by this class of inhibitors, although at a reduced potency compared to HPV6 E1. This latter finding raises the possibility that this class of small molecule inhibitors could be optimized for activity against many HPV types [94White PW, Faucher AM, Massariol MJ, et al. Biphenylsulfonacetic acid inhibitors of the human papillomavirus type 6 E1 helicase inhibit ATP hydrolysis by an allosteric mechanism involving tyrosine 486 Antimicrob Agents Chemother 2005; 49: 4834-2.]. Using the crystal structure of the HPV18 E1 helicase domain as a tool to determine the possible binding region of these inhibitors revealed that Tyr-486 in HPV6 E1 is located close to the highly conserved Lys-484, an essential residue for ATP interaction and catalysis [95Abbate EA, Berger JM, Botchan MR. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2 Genes Dev 2004; 18: 1981-96.] (Fig. 3B, C). It has therefore been speculated that the interaction of these inhibitors with Tyr-486 may induce a conformational change in E1 that would indirectly affect ATP binding, accounting for their allosteric mode of action. Consistent with the fact that ATP hydrolysis is required for helicase activity, the inhibitors also inhibited the helicase activity of HPV6 E1 in vitro. Despite these promising results, the biphenyl-sulfonacetic acid inhibitors were not active in cell-based assays [93Faucher AM, White PW, Brochu C, et al. Discovery of small-molecule inhibitors of the ATPase activity of human papillomavirus E1 helicase J Med Chem 2004; 47: 18-21.], perhaps attributable in part to the relatively high intracellular concentration of ATP. Nonetheless, this class of inhibitors has demonstrated the potential of the E1 ATPase domain as a target for inhibition of HPV DNA replication by small molecules. The identification of the small molecule binding pocket on the HPV E1 protein by X-ray crystallo-graphy of an E1-inhibitor complex would be the next logical step towards the design of second generation inhibitors with increased specificity and activity.
E1-E2 INTERACTION INHIBITORS
To date, relatively few small molecule inhibitors of protein-protein interactions have been identified. A potential explanation for this is that small molecules may have difficulty in competing against the large surface area typically involved in a protein-protein interface, which furthermore, tend to be fairly flat and devoid of small molecule binding pockets. Nevertheless, several studies have demonstrated that substitution of a single critical residue at an interface is sufficient to greatly weaken an interaction [96Cunningham BC, Wells JA. Comparison of a structural and a functional epitope J Mol Biol 1993; 234: 554-63.], thereby raising the possibility that some protein interfaces might be amenable to small molecule inhibition. For HPV, one such protein-protein interaction target includes the binding between E1 and E2. Viral DNA replication is initiated by the recruitment of E1, by E2, to specific DNA sequences within the viral origin of replication. Formation of a ternary complex between E1, E2, and the origin facilitates recognition of the viral origin by E1 and serves as a platform for the recruitment of additional E1 molecules and their assembly into replication-competent double hexamers capable of bidirectional unwinding [29Titolo S, Pelletier A, Pulichino AM, et al. Identification of domains of the human papillomavirus type 11 E1 helicase involved in oligomerization and binding to the viral origin J Virol 2000; 74: 7349-61., 97Fouts ET, Yu X, Egelman EH, Botchan MR. Biochemical and electron microscopic image analysis of the hexameric E1 helicase J Biol Chem 1999; 274: 4447-58., 98Sedman J, Stenlund A. The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities J Virol 1998; 72: 6893-7.]. Assembly of this E1-E2-ori ternary complex is dependent upon the interaction of E1 and E2 with DNA, and upon a critical protein interaction between the TAD of E2 and the C-terminal ATPase domain of E1 [42Berg M, Stenlund A. Functional interactions between papillomavirus E1 and E2 proteins J Virol 1997; 71: 3853-63.-44Bonne-Andrea C, Tillier F, McShan GD, Wilson VG, Clertant P. Bovine papillomavirus type 1 DNA replication: the transcriptional activator E2 acts in vitro as a specificity factor J Virol 1997; 71: 6805-15., 46Mohr IJ, Clark R, Sun S, et al. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator Science 1990; 250: 1694-9.-49Yang L, Li R, Mohr IJ, Clark R, Botchan MR. Activation of BPV-1 replication in vitro by the transcription factor E2 Nature 1991; 353: 628-32.]. A priori, any of the protein-protein or protein-DNA interactions that take place at the origin can be targeted for the development of small molecule inhibitors for the treatment of HPV lesions. To date, substantial efforts have been made toward the discovery of compounds that specifically target and inhibit the DNA-binding activities of E1 and E2 or prevent the E1-E2 interaction, with only a few studies demonstrating efficacy in vivo. A class of small molecules termed indandiones, which specifically inhibit the E1-E2 protein interaction, have been identified as the first small molecule inhibitors of HPV DNA replication [99Yoakim C, Ogilvie WW, Goudreau N, et al. Discovery of the first series of inhibitors of human papillomavirus type 11: inhibition of the assembly of the E1-E2-Origin DNA complex Bioorg Med Chem Lett 2003; 13: 2539-41., 100White PW, Titolo S, Brault K, et al. Inhibition of human papillomavirus DNA replication by small molecule antagonists of the E1-E2 protein interaction J Biol Chem 2003; 278: 26765-72.]. The original inhibitor in this class of compounds was identified by high-throughput screening of over 100,000 diverse chemical compounds for its ability to interfere with the cooperative binding of recombinant HPV11 E1 and E2 to radiolabeled origin DNA. Medicinal chemistry efforts then led to the synthesis of more active analogs capable of inhibiting the assembly of the HPV11 E1-E2-origin DNA complex with low nanomolar potency in vitro and with activity in cell-based assays [100White PW, Titolo S, Brault K, et al. Inhibition of human papillomavirus DNA replication by small molecule antagonists of the E1-E2 protein interaction J Biol Chem 2003; 278: 26765-72.] (Fig. 4A ). This set of inhibitors featured an indandione system spirofused onto a substituted tetrahydrofuran ring and was determined to act by binding to the TAD domain of E2, the same region of the protein that interacts with E1. Crystal structures of the HPV11 E2 TAD alone and in complex with an indandione inhibitor provided a detailed understanding of the mechanism by which the indandione inhibitors bind to E2 to disrupt the E1-E2 interaction [101Wang Y, Coulombe R, Cameron DR, et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor J Biol Chem 2004; 279: 6976-85.] (Fig. 4B, C). Comparison of the two structures revealed that inhibitor binding did not significantly alter the protein backbone but did induce significant movement of several amino acid side chains at the binding site. These changes in side chain conformation, particularly of residues Tyr-19, His-32, Leu-94, and Glu-100, resulted in the formation of a deep hydrophobic pocket that tightly binds to the indandione moiety of the inhibitor. The structure also revealed that the carboxylate moiety of the inhibitor, known to be important for potency, forms hydrogen bonds with amides from the protein backbone. These inhibitors were determined to bind specifically to the E2 protein of the two most prevalent low-risk HPV types, HPV6 and -11, rather than the high-risk types [100White PW, Titolo S, Brault K, et al. Inhibition of human papillomavirus DNA replication by small molecule antagonists of the E1-E2 protein interaction J Biol Chem 2003; 278: 26765-72.]. This lack of inhibition of high-risk E2 was attributable in part to residue His-32, which is a tyrosine in the E2 of most high-risk HPV types. Substitution of His-32 for tyrosine in HPV11 E2 was found to abrogate inhibitor binding, although it did not affect interaction with E1. In fact, mutational analysis of several residues in or near the inhibitor binding pocket provided functional evidence for specific interactions between Tyr-19 and E1, and between His-32 and the inhibitor [101Wang Y, Coulombe R, Cameron DR, et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor J Biol Chem 2004; 279: 6976-85.]. Substitution of His-32 is predicted to prevent the formation of the deep hydrophobic inhibitor binding pocket. Other subtle conformational changes may also explain why the indandione class of inhibitors bind to HPV6 E2 less tightly than to HPV11 E2. Notably, mutation of Glu-100 to alanine in HPV11 E2 was also determined to increase the sensitivity of E2 to the indandione inhibitors by 10-fold in vitro [102Fradet-Turcotte A, Archambault J. Recent advances in the search for antiviral agents against human papillomaviruses Antivir Ther 2007; 12: 431-51.]. A subsequent screen also led to the discovery and optimization of a second series of reversible E1-E2 interaction inhibitors (Fig. 4A). Modeling studies suggested that these repaglinide-derived inhibitors form weaker interactions with the E2 TAD but occupy a larger portion of the inhibitor binding pocket compared to the indandione series. The fact that potent repaglinide derivatives, with IC50 values in the nanomolar range against HPV6 and HPV11 E2, could be obtained after only a few rounds of optimization makes this series a promising avenue for the development of small molecule inhibitors of the E1-E2 interaction [103White PW, Faucher AM, Goudreau N. Small molecule inhibitors of the human papillomavirus e1-e2 interaction Curr Top Microbiol Immunol 2011; 348: 61-88.]. Of interest is the finding that E1 and E2 interact through a relatively large surface area of approximately 950 Å2, which includes the inhibitor binding pocket [95Abbate EA, Berger JM, Botchan MR. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2 Genes Dev 2004; 18: 1981-96.]. Thus, the indandione and repaglinide-derived inhibitors provide tangible examples that small molecules can indeed inhibit relatively large protein-protein interfaces.
). This set of inhibitors featured an indandione system spirofused onto a substituted tetrahydrofuran ring and was determined to act by binding to the TAD domain of E2, the same region of the protein that interacts with E1. Crystal structures of the HPV11 E2 TAD alone and in complex with an indandione inhibitor provided a detailed understanding of the mechanism by which the indandione inhibitors bind to E2 to disrupt the E1-E2 interaction [101Wang Y, Coulombe R, Cameron DR, et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor J Biol Chem 2004; 279: 6976-85.] (Fig. 4B, C). Comparison of the two structures revealed that inhibitor binding did not significantly alter the protein backbone but did induce significant movement of several amino acid side chains at the binding site. These changes in side chain conformation, particularly of residues Tyr-19, His-32, Leu-94, and Glu-100, resulted in the formation of a deep hydrophobic pocket that tightly binds to the indandione moiety of the inhibitor. The structure also revealed that the carboxylate moiety of the inhibitor, known to be important for potency, forms hydrogen bonds with amides from the protein backbone. These inhibitors were determined to bind specifically to the E2 protein of the two most prevalent low-risk HPV types, HPV6 and -11, rather than the high-risk types [100White PW, Titolo S, Brault K, et al. Inhibition of human papillomavirus DNA replication by small molecule antagonists of the E1-E2 protein interaction J Biol Chem 2003; 278: 26765-72.]. This lack of inhibition of high-risk E2 was attributable in part to residue His-32, which is a tyrosine in the E2 of most high-risk HPV types. Substitution of His-32 for tyrosine in HPV11 E2 was found to abrogate inhibitor binding, although it did not affect interaction with E1. In fact, mutational analysis of several residues in or near the inhibitor binding pocket provided functional evidence for specific interactions between Tyr-19 and E1, and between His-32 and the inhibitor [101Wang Y, Coulombe R, Cameron DR, et al. Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor J Biol Chem 2004; 279: 6976-85.]. Substitution of His-32 is predicted to prevent the formation of the deep hydrophobic inhibitor binding pocket. Other subtle conformational changes may also explain why the indandione class of inhibitors bind to HPV6 E2 less tightly than to HPV11 E2. Notably, mutation of Glu-100 to alanine in HPV11 E2 was also determined to increase the sensitivity of E2 to the indandione inhibitors by 10-fold in vitro [102Fradet-Turcotte A, Archambault J. Recent advances in the search for antiviral agents against human papillomaviruses Antivir Ther 2007; 12: 431-51.]. A subsequent screen also led to the discovery and optimization of a second series of reversible E1-E2 interaction inhibitors (Fig. 4A). Modeling studies suggested that these repaglinide-derived inhibitors form weaker interactions with the E2 TAD but occupy a larger portion of the inhibitor binding pocket compared to the indandione series. The fact that potent repaglinide derivatives, with IC50 values in the nanomolar range against HPV6 and HPV11 E2, could be obtained after only a few rounds of optimization makes this series a promising avenue for the development of small molecule inhibitors of the E1-E2 interaction [103White PW, Faucher AM, Goudreau N. Small molecule inhibitors of the human papillomavirus e1-e2 interaction Curr Top Microbiol Immunol 2011; 348: 61-88.]. Of interest is the finding that E1 and E2 interact through a relatively large surface area of approximately 950 Å2, which includes the inhibitor binding pocket [95Abbate EA, Berger JM, Botchan MR. The X-ray structure of the papillomavirus helicase in complex with its molecular matchmaker E2 Genes Dev 2004; 18: 1981-96.]. Thus, the indandione and repaglinide-derived inhibitors provide tangible examples that small molecules can indeed inhibit relatively large protein-protein interfaces.
The ability of the indandione compounds to inhibit HPV genome replication in vivo, especially that of low-risk HPV types, demonstrates for the first time the therapeutic potential of the E2 protein as a target for the development of small molecule inhibitors of HPV protein interactions, particularly in this case for the treatment of anogenital warts caused by HPV6 and -11. More specifically, the localization and characterization of this inhibitor binding pocket in the E2 TAD provides a potential new therapeutic target for the treatment of HPV infections. These studies have laid the foundation for the use of structure-based approaches to rationally design or virtually screen inhibitory compounds that are capable of binding to all or part of this identified inhibitor binding pocket. An understanding of such associations will aid in the design of additional small molecules that will exhibit improved biological effects and favorable associations with the E2 proteins of a wider range of HPV types. Given that the E1-E2 interaction is highly conserved between mucosal and cutaneous viruses, it is possible that antiviral compounds that antagonize this interaction would be effective against infections caused by many different HPV types.
While inhibition of E1 and/or E2 function represents a valuable strategy for the treatment of HPV-associated benign lesions such as warts, in which the viral genome is maintained as an episome, it is important to realize that expression of these two viral proteins is often lost during the process of malignant transformation. Integration of the viral DNA into the host genome and the concomitant loss of the E2 protein is a well-established risk factor for cancer development, as it often results in increased expression of the E6 and E7 oncogenes, albeit not invariably [104Hafner N, Driesch C, Gajda M, et al. Integration of the HPV16 genome does not invariably result in high levels of viral oncogene transcripts Oncogene 2008; 27: 1610-7.]. Since both E6 and E7 were shown to be essential for the proliferation and survival of cervical carcinoma cells, they are in principle the targets of choice for the treatment of HPV-induced cancers.
TARGETING THE E6 AND E7 ONCOPROTEINS
The molecular functions of the HPV E6 and E7 oncoproteins involved in cellular transformation and immortalization represent validated targets for therapeutic intervention. Both are small, zinc finger-containing proteins that need to be continually expressed for progression and maintenance of HPV-positive cancers. As mentioned above, expression of E6 and E7 is transcriptionally regulated by E2 during the normal viral life cycle. In cancer progression, however, integration of the viral genome frequently occurs, often in a manner that disrupts the E2 open reading frame and eliminates the E2-mediated transcriptional control of the early region. This results in constitutive and/or increased overexpression of E6 and E7 in HPV-associated cancers [104Hafner N, Driesch C, Gajda M, et al. Integration of the HPV16 genome does not invariably result in high levels of viral oncogene transcripts Oncogene 2008; 27: 1610-7.]. Both E6 and E7 allow for the efficient immortalization of infected keratinocytes and thereby play an important role in HPV-induced carcinogenesis. In fact, cell culture and animal model studies have clearly established that expression of E6 and E7 from high-risk HPV types is necessary and sufficient to immortalize primary keratinocytes [105DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells J Virol 2003; 77: 1551-63., 106Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes EMBO J 1989; 8: 3905-10.], induce epithelial cell hyperplasia [107Herber R, Liem A, Pitot H, Lambert PF. Squamous epithelial hyperplasia and carcinoma in mice transgenic for the human papillomavirus type 16 E7 oncogene J Virol 1996; 70: 1873-81., 108Song S, Pitot HC, Lambert PF. The human papillomavirus type 16 E6 gene alone is sufficient to induce carcinomas in transgenic animals J Virol 1999; 73: 5887-93.], abrogate DNA damage responses [109Song S, Gulliver GA, Lambert PF. Human papillomavirus type 16 E6 and E7 oncogenes abrogate radiation-induced DNA damage responses in vivo through p53-dependent and p53-independent pathways Proc Natl Acad Sci USA 1998; 95: 2290-5., 110Demers GW, Halbert CL, Galloway DA. Elevated wild-type p53 protein levels in human epithelial cell lines immortalized by the human papillomavirus type 16 E7 gene Virology 1994; 198: 169-74.], and induce genomic instability [111Schaeffer AJ, Nguyen M, Liem A, et al. E6 and E7 oncoproteins induce distinct patterns of chromosomal aneuploidy in skin tumors from transgenic mice Cancer Res 2004; 64: 538-46., 112White AE, Livanos EM, Tlsty TD. Differential disruption of genomic integrity and cell cycle regulation in normal human fibroblasts by the HPV oncoproteins Genes Dev 1994; 8: 666-77.]. The oncogenic potential of these proteins is mediated by specific interactions with key cell cycle regulatory proteins, namely those of E6 with the tumor suppressor protein p53, and of E7 with the retinoblastoma (Rb) family of pocket proteins. One important function of p53 is to transactivate the expression of various regulators that induce cell cycle arrest in G1/S and/or apoptosis in response to DNA damage, while that of Rb and the related pocket proteins p107 and p130 is to control the transition at the G1/S phase of the cell cycle by binding and regulating the activity of the E2F family of transcription factors. Consequently, through these interactions, E7 stimulates quiescent cells to re-enter S-phase, while E6 prevents cellular growth arrest or DNA damage-induced apoptosis [27Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity Rev Med Virol 2006; 16: 83-97.] (Fig. 5A ). In support of this, studies have shown that an increase in cell proliferation was observed when the HPV16 E7 gene was expressed in the lens of transgenic mice or in the basal epithelial cells of the mouse epidermis [113Pan H, Griep AE. Altered cell cycle regulation in the lens of HPV-16 E6 or E7 transgenic mice: implications for tumor suppressor gene function in development Genes Dev 1994; 8: 1285-99., 114Riley RR, Duensing S, Brake T, et al. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis Cancer Res 2003; 63: 4862-71.]. In other studies, repression of E6 and/or E7 expression in cervical carcinoma cell lines, by transfection of a functional E2 or by siRNAs, restored the p53 and Rb pathways and induced cellular senescence [105DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells J Virol 2003; 77: 1551-63., 115Jiang M, Milner J. Selective silencing of viral gene expression in HPV-positive human cervical carcinoma cells treated with siRNA, a primer of RNA interference Oncogene 2002; 21: 6041-8.-119Hall AH, Alexander KA. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells J Virol 2003; 77: 6066-9.]. Moreover, it has been suggested that both E6 and E7 utilize the ubiquitin-proteosome system to target these cellular proteins for degradation and inactivation [120Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway Cancer Res 1996; 56: 4620-., 121Huh K, Zhou X, Hayakawa H, et al. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor J Virol 2007; 81: 9737-47.]. This has been first demonstrated for E6, which associates with the cellular E3 ubiquitin ligase E6-associated protein (E6AP), forming an E6/E6AP complex capable of binding p53 and inducing its specific ubiquitination and subsequent degradation by the proteasome [122Scheffner M, Whitaker NJ. Human papillomavirus-induced carcinogenesis and the ubiquitin-proteasome system Semin Cancer Biol 2003; 13: 59-67.]. Notably, the E6 protein of low-risk HPV types does not induce p53 degradation correlating with their weak transformation potential. HPV31 viral genomes containing mutations in E6 that reduce E6AP binding or prevent p53 interaction were incapable of stable episomal maintenance and immortalization of primary keratinocytes [123Thomas JT, Hubert WG, Ruesch MN, Laimins LA. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes Proc Natl Acad Sci USA 1999; 96: 8449-54.]. Similar results were obtained with HPV31 viral genomes containing mutant E7 genes [123Thomas JT, Hubert WG, Ruesch MN, Laimins LA. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes Proc Natl Acad Sci USA 1999; 96: 8449-54.]. Together, these and other findings (reviewed in [27Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity Rev Med Virol 2006; 16: 83-97.]) suggest that preventing the p53 degradation function of E6 or the interaction between E7 and Rb and related pocket proteins represent promising avenues for the discovery of small molecule inhibitors of HPV-induced carcinogenesis.
). In support of this, studies have shown that an increase in cell proliferation was observed when the HPV16 E7 gene was expressed in the lens of transgenic mice or in the basal epithelial cells of the mouse epidermis [113Pan H, Griep AE. Altered cell cycle regulation in the lens of HPV-16 E6 or E7 transgenic mice: implications for tumor suppressor gene function in development Genes Dev 1994; 8: 1285-99., 114Riley RR, Duensing S, Brake T, et al. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis Cancer Res 2003; 63: 4862-71.]. In other studies, repression of E6 and/or E7 expression in cervical carcinoma cell lines, by transfection of a functional E2 or by siRNAs, restored the p53 and Rb pathways and induced cellular senescence [105DeFilippis RA, Goodwin EC, Wu L, DiMaio D. Endogenous human papillomavirus E6 and E7 proteins differentially regulate proliferation, senescence, and apoptosis in HeLa cervical carcinoma cells J Virol 2003; 77: 1551-63., 115Jiang M, Milner J. Selective silencing of viral gene expression in HPV-positive human cervical carcinoma cells treated with siRNA, a primer of RNA interference Oncogene 2002; 21: 6041-8.-119Hall AH, Alexander KA. RNA interference of human papillomavirus type 18 E6 and E7 induces senescence in HeLa cells J Virol 2003; 77: 6066-9.]. Moreover, it has been suggested that both E6 and E7 utilize the ubiquitin-proteosome system to target these cellular proteins for degradation and inactivation [120Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway Cancer Res 1996; 56: 4620-., 121Huh K, Zhou X, Hayakawa H, et al. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor J Virol 2007; 81: 9737-47.]. This has been first demonstrated for E6, which associates with the cellular E3 ubiquitin ligase E6-associated protein (E6AP), forming an E6/E6AP complex capable of binding p53 and inducing its specific ubiquitination and subsequent degradation by the proteasome [122Scheffner M, Whitaker NJ. Human papillomavirus-induced carcinogenesis and the ubiquitin-proteasome system Semin Cancer Biol 2003; 13: 59-67.]. Notably, the E6 protein of low-risk HPV types does not induce p53 degradation correlating with their weak transformation potential. HPV31 viral genomes containing mutations in E6 that reduce E6AP binding or prevent p53 interaction were incapable of stable episomal maintenance and immortalization of primary keratinocytes [123Thomas JT, Hubert WG, Ruesch MN, Laimins LA. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes Proc Natl Acad Sci USA 1999; 96: 8449-54.]. Similar results were obtained with HPV31 viral genomes containing mutant E7 genes [123Thomas JT, Hubert WG, Ruesch MN, Laimins LA. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes Proc Natl Acad Sci USA 1999; 96: 8449-54.]. Together, these and other findings (reviewed in [27Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity Rev Med Virol 2006; 16: 83-97.]) suggest that preventing the p53 degradation function of E6 or the interaction between E7 and Rb and related pocket proteins represent promising avenues for the discovery of small molecule inhibitors of HPV-induced carcinogenesis.
In addition to p53 and Rb, E6 and E7 have also been shown to interact with many other cellular proteins (recently reviewed in [124Wise-Draper TM, Wells SI. Papillomavirus E6 and E7 proteins and their cellular targets Front Biosci 2008; 13: 1003-7.]). For instance, high-risk HPV E6 proteins containing the C-terminal sequence X-T/S-X-V/L interact with the PDZ-domain containing proteins hDlg, MUPP1, hScribble, MAGI-1-3, and PTPN13 targeting them for proteasome-mediated degradation [125Massimi P, Gammoh N, Thomas M, Banks L. HPV E6 specifically targets different cellular pools of its PDZ domain-containing tumour suppressor substrates for proteasome-mediated degradation Oncogene 2004; 23: 8033-9.-130Spanos WC, Hoover A, Harris GF, et al. The PDZ binding motif of human papillomavirus type 16 E6 induces PTPN13 loss, which allows anchorage-independent growth and synergizes with ras for invasive growth J Virol 2008; 82: 2493-500.]. These cellular proteins, many of which are tumor suppressors [125Massimi P, Gammoh N, Thomas M, Banks L. HPV E6 specifically targets different cellular pools of its PDZ domain-containing tumour suppressor substrates for proteasome-mediated degradation Oncogene 2004; 23: 8033-9., 131Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors Science 2000; 289: 113-6., 132Borg JP. hScrib: a potential novel tumor suppressor Pathol Biol (Paris) 2004; 52: 328-1.], function as structural organizers of signaling molecules in diverse cellular compartments to modulate cell polarity, cell-cell adhesion, ion transport, and normal cell growth. E6 mediates the degradation of PDZ-domain proteins through both E6AP-dependent and E6AP-independent mechanisms [133Kuballa P, Matentzoglu K, Scheffner M. The role of the ubiquitin ligase E6-AP in human papillomavirus E6-mediated degradation of PDZ domain-containing proteins J Biol Chem 2007; 282: 65-71.-135Ainsworth J, Thomas M, Banks L, Coutlee F, Matlashewski G. Comparison of p53 and the PDZ domain containing protein MAGI-3 regulation by the E6 protein from high-risk human papillomaviruses J Virol 2008; 5: 67.]. This degradation results in the disruption of multiple signaling pathways and has been shown to be necessary for cell transformation [136Simonson SJ, Difilippantonio MJ, Lambert PF. Two distinct activities contribute to human papillomavirus 16 E6's oncogenic potential Cancer Res 2005; 65: 8266-73.], suggesting that abrogation of PDZ-domain containing proteins contributes to the oncogenic potential of E6. Studies of transgenic mice provide further support for this notion by demonstrating that an increase in cellular proliferation and induction of epithelial hyperplasia in the mouse lens and in the epidermis is dependent upon the binding of E6 to PDZ-domain proteins [137Nguyen MM, Nguyen ML, Caruana G, et al. Requirement of PDZ-containing proteins for cell cycle regulation and differentiation in the mouse lens epithelium Mol Cell Biol 2003; 23: 8970-1., 138Nguyen ML, Nguyen MM, Lee D, Griep AE, Lambert PF. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6's induction of epithelial hyperplasia in vivo J Virol 2003; 77: 6957-4.]. Interfering with these processes could ultimately be a valuable approach for the development of specific HPV antiviral compounds. In fact, the efficacy of this approach has been shown by the use of synthetic peptides which efficiently abolished the ability of E6 to promote the degradation of the PDZ-domain proteins hDlg and MAGI-1 in vitro [139Sterlinko GH, Weber M, Elston R, et al. Inhibition of E6-induced degradation of its cellular substrates by novel blocking peptides J Mol Biol 2004; 335: 971-85.]. In addition to its interaction with the Rb family of proteins, increasing evidence suggests that E7 influences E2F-induced transcription through interactions with several other cellular proteins including the class I histone deacetylases (HDAC) [140Brehm A, Nielsen SJ, Miska EA, et al. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth EMBO J 1999; 18: 2449-58., 141Longworth MS, Wilson R, Laimins LA. HPV31 E7 facilitates replication by activating E2F2 transcription through its interaction with HDACs EMBO J 2005; 24: 1821-30.]. HDAC activity has been associated with cancer and to date, several HDAC inhibitors are in clinical trials [142Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future Nat Rev Drug Discov 2006; 5: 37-50.]. The significance of the interaction between E7 and HDAC was demonstrated in reverse genetic experiments, where HPV31 genomes containing mutations in the E7 HDAC binding sequences were severely impaired in mediating episomal maintenance and in modulating cellular proliferation of primary keratinocytes [143Longworth MS, Laimins LA. The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31 J Virol 2004; 78: 3533-41.]. These results validate this protein interaction as a potential antiviral target. Despite being attractive molecular targets from a biological standpoint, E6 and E7 have historically proven to be refractory to inhibition by small molecules, perhaps because of their small size and/or lack of inhibitor binding pockets on their surface. Nevertheless, some important progress was recently reported toward the development of E6 inhibitors and is summarized below.
E6-E6AP INHIBITORS
A key mechanism utilized by papillomaviruses to prevent apoptosis of infected cells involves the degradation of the tumor suppressor protein p53 by E6, in association with the cellular ubiquitin ligase E6AP, thereby also contributing to the immortalization abilities of high-risk HPV E6 proteins. Although all HPV E6 proteins have been shown to bind to p53 [144Li X, Coffino P. High-risk human papillomavirus E6 protein has two distinct binding sites within p53, of which only one determines degradation J Virol 1996; 70: 4509-16.], degradation of p53 is specific to high-risk HPV types and relies upon the interaction between E6 and E6AP [145Huibregtse JM, Scheffner M, Howley PM. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53 Mol Cell Biol 1993; 13: 775-84.]. It has been demonstrated that downregulation of E6AP expression by antisense olignucleotides [146Beer-Romero P, Glass S, Rolfe M. Antisense targeting of E6AP elevates p53 in HPV-infected cells but not in normal cells Oncogene 1997; 14: 595-602.] or RNA interference [147Hengstermann A, D'Silva MA, Kuballa P, et al. Growth suppression induced by downregulation of E6-AP expression in human papillomavirus-positive cancer cell lines depends on p53 J Virol 2005; 79: 9296-300.], or inhibition of E6AP by dominant negative mutant proteins [148Talis AL, Huibregtse JM, Howley PM. The role of E6AP in the regulation of p53 protein levels in human papillomavirus (HPV)-positive and HPV-negative cells J Biol Chem 1998; 273: 6439-45.] resulted in accumulation of p53 protein levels and growth suppression in HPV-positive but not HPV-negative cells. Together, E6 and E6AP bind within the core DNA-binding domain of p53 and facilitate its ubiquitination and proteasomal degradation. The essential role of the E6/E6AP complex in the degradation of p53 is further supported by studies which demonstrated that mutant p53 proteins unable to bind E6 were not susceptible to E6AP-mediated degradation [149Scheffner M, Takahashi T, Huibregtse JM, Minna JD, Howley PM. Interaction of the human papillomavirus type 16 E6 oncoprotein with wild-type and mutant human p53 proteins J Virol 1992; 66: 5100-.], while E6AP mutants defective for E6 binding were unable to interact with p53 [150Huibregtse JM, Scheffner M, Howley PM. Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins Mol Cell Biol 1993; 13: 4918-27.]. As such, inhibition of the E6-E6AP complex represents an attractive therapeutic target, as it would ultimately re-activate p53 function in high-risk HPV-positive cells leading to growth arrest and apoptosis of HPV-transformed cells.
Different approaches have been tested to design effective inhibitors of the E6-E6AP complex. The starting point of one approach was the identification, by sequence alignment of several E6 interacting proteins, including E6AP, E6BP, paxillin, and interferon-regulatory factor 3 (IRF3), of a consensus E6-binding motif, L-x-x-Ф-L-s-h, where L is leucine, xx is a dipeptide in which one of the residues is aspartate, asparagine, glutamine, or glutamate, Ф is a hydrophobic residue (usually leucine), s is a small amino acid (glycine or alanine), and h is usually aspartate, asparagine, glutamine, or glutamate [151Baleja JD, Cherry JJ, Liu Z, et al. Identification of inhibitors to papillomavirus type 16 E6 protein based on three-dimensional structures of interacting proteins Antiviral Res 2006; 72: 49-59.]. This motif folds into an α-helix, such that the three leucine residues form a hydrophobic surface on one side of the helix and the charged amino acids lie on the opposite side. Mutagenesis studies revealed the critical importance of the three leucine residues (Leu-9, Leu-12 and Leu-13) for E6 binding [152Be X, Hong Y, Wei J, et al. Solution structure determination and mutational analysis of the papillomavirus E6 interacting peptide of E6AP Biochemistry 2001; 40: 1293-9.]. Substitution of any leucine with alanine in the binding motif was shown to abolish binding to E6, whereas mutations of polar residues on the opposite side of the helix were shown to weaken binding. These features led to the suggestion that E6AP interacts with E6 through a “charged leucine” binding motif [151Baleja JD, Cherry JJ, Liu Z, et al. Identification of inhibitors to papillomavirus type 16 E6 protein based on three-dimensional structures of interacting proteins Antiviral Res 2006; 72: 49-59., 152Be X, Hong Y, Wei J, et al. Solution structure determination and mutational analysis of the papillomavirus E6 interacting peptide of E6AP Biochemistry 2001; 40: 1293-9.] (Fig. 5B). As such, two small stable peptides were designed, where the minimal E6-binding consensus sequence was grafted into the single α-helix of the third zinc-finger of Sp1 and the Trp-cage peptide [153Liu Y, Liu Z, Androphy E, Chen J, Baleja JD. Design and characterization of helical peptides that inhibit the E6 protein of papillomavirus Biochemistry 2004; 43: 7421-31.]. Unfortunately, these peptides were only weak competitors of the interaction between E6-E6AP in comparison with the 18-mer peptide from E6AP (residues 391 to 408) that encompasses the complete E6-binding motif and inhibits binding of E6AP to HPV16 E6 with an IC50 of 10 µM in vitro [139Sterlinko GH, Weber M, Elston R, et al. Inhibition of E6-induced degradation of its cellular substrates by novel blocking peptides J Mol Biol 2004; 335: 971-85., 153Liu Y, Liu Z, Androphy E, Chen J, Baleja JD. Design and characterization of helical peptides that inhibit the E6 protein of papillomavirus Biochemistry 2004; 43: 7421-31.]. Structure of this 18-mer peptide lead was obtained by NMR [152Be X, Hong Y, Wei J, et al. Solution structure determination and mutational analysis of the papillomavirus E6 interacting peptide of E6AP Biochemistry 2001; 40: 1293-9.] and is shown in Fig. (5C). Based on these structural and functional studies of E6-binding proteins, Baleja et al. [151Baleja JD, Cherry JJ, Liu Z, et al. Identification of inhibitors to papillomavirus type 16 E6 protein based on three-dimensional structures of interacting proteins Antiviral Res 2006; 72: 49-59.] constructed an E6-binding pharmacophore model which they then used to identify potential inhibitory molecules by virtual screening of the National Cancer Institute (NCI) chemical database (approximately 240, 000 compounds) and Sigma Aldrich chemical database (97, 000 compounds). 73 candidate compounds were identified, which were further tested in vitro using a p53 degradation assay and a gel-based E6-E6AP binding assay. Selectivity and cell permeability were also tested. 5 lead candidates were identified, which displayed acceptable selectivity and inhibitory activity (IC50 values ranging between 17 µM-29 µM), however, no structure-activity relationship could be established from this limited dataset. Overall, these compounds, especially Compound 9, provide an exciting starting point for the synthesis of future inhibitors with improved potency and specificity (Fig. 5D). Future studies aimed at determining the structure of HPV E6-inhibitor complexes will ultimately increase our current understanding of how these compounds interact with E6 and will facilitate the identification of a potential inhibitor binding interface on E6. To date, no such interface has been mapped, although it has been suggested that the binding pocket for E6AP lies at the junction of both zinc-binding domains [151Baleja JD, Cherry JJ, Liu Z, et al. Identification of inhibitors to papillomavirus type 16 E6 protein based on three-dimensional structures of interacting proteins Antiviral Res 2006; 72: 49-59.]. The reported three-dimensional structural analysis of the C-terminal zinc-finger domain of HPV16 E6 provides a tool to assist with these studies [154Nomine Y, Masson M, Charbonnier S, et al. Structural and functional analysis of E6 oncoprotein: insights in the molecular pathways of human papillomavirus-mediated pathogenesis Mol Cell 2006; 21: 665-78.]. The identification of small synthetic peptides that specifically bind to and inhibit the oncogenic activity of E6 may also provide valuable information defining critical amino acid residues and contact points [155Dymalla S, Scheffner M, Weber E, et al. A novel peptide motif binding to and blocking the intracellular activity of the human papillomavirus E6 oncoprotein J Mol Med 2009; 87: 321-1.]. Overall, the fact that E6AP appears to be the primary protein needed for the E6-mediated degradation of p53, PDZ-domain containing proteins, and possibly other host proteins, strengthens the idea that obstruction of the E6AP-binding pocket on E6 is an attractive strategy for the development of small molecule inhibitors against HPV-induced pathogenesis.
CONCLUSIONS
Modulating protein-protein interactions is becoming an increasingly attractive strategy for the development of small molecule inhibitors, but remains a challenge due to various obstacles such as lack of well-defined binding pockets and the need to compete with large protein-protein binding interfaces, etc. Nevertheless, reports on the discovery of inhibitors with nanomolar affinities and high specificities for their targets are increasing, demonstrating the feasibility of this approach for the treatment of diseases. Tremendous progress has been made in increasing our knowledge of the function of many of the viral proteins involved in the HPV life cycle. Among these proteins are E1, E2, E6, and E7, all of which have been validated as essential for viral pathogenesis and thus represent genuine targets for small molecule-based approaches for the treatment of HPV-associated diseases. As shown above, small molecules that inhibit specific functions of these viral proteins are now emerging and have provided pharmacological validation that these viral targets are druggable. In particular, two protein-protein interactions have been validated as HPV targets by small molecule inhibitors, namely the E1-E2 interaction and the E6-E6AP interaction. The use of small molecule-based approaches to interfere with the binding of E2 to Brd4 also represents an unexplored target for the treatment of HPV infections. Targeting this interaction is substantiated by a study reporting the development of a peptide that consists of the last 20 amino acids of Brd4 that is able to interact with E2 and specifically interfere with its ability to tether the viral DNA to chromatin [156Abbate EA, Voitenleitner C, Botchan MR. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association Mol Cell 2006; 24: 877-9.]. Another yet unexplored target is the interaction between E6 and PDZ-domain containing proteins. PDZ domains have well-defined binding sites, making them promising targets for drug discovery. Additional studies clarifying the function of these domains in normal and infected cells, however, are necessary before PDZ interactions can be targeted for drug development. Alternatively, the development of small molecules that target p53 and protect it from E6-mediated degradation, rather than inhibit E6 directly, represents a promising novel strategy for the treatment of HPV-associated cancers. The recent identification of the small molecule inhibitor RITA serves as an example. RITA was determined to efficiently suppress the growth of cervical cancer cells both in vitro and in vivo. The molecule was found to target p53 directly and prevent its interaction with E6AP to protect it from proteasomal E6-mediated degradation in both HPV16- and HPV18-infected cells [157Zhao CY, Szekely L, Bao W, Selivanova G. Rescue of p53 function by small-molecule RITA in cervical carcinoma by blocking E6-mediated degradation Cancer Res 2010; 70: 3372-81.]. The recent screening of a chemical library of compounds also identified two small molecules that selectively caused growth suppression of cervical cancer cells. Although not clearly established, inhibition of p53 interaction with E6 or E6AP is considered as a possible mechanism of action [158Kochetkov DV, Il'inskaia GV, Komarov PG, et al. Transcriptional inhibition of human papilloma virus in cervical carcinoma cells reactivates functions of the tumor suppressor p53 Mol Biol (Mosk) 2007; 41: 515-23.]. Importantly, the identification of small peptides that bind to E6 and interfere with the E6-mediated degradation of p53 or with other additional tumor relevant E6 interactions may also serve as a starting point for the development of small molecule peptide mimetics against HPV infection [155Dymalla S, Scheffner M, Weber E, et al. A novel peptide motif binding to and blocking the intracellular activity of the human papillomavirus E6 oncoprotein J Mol Med 2009; 87: 321-1.]. Finally, viruses have been shown in recent years to alter cellular physiology by modulating the expression of small noncoding regulatory RNAs or microRNAs (miRNAs or miR) [159Hawkins SM, Buchold GM, Matzuk MM. Minireview: the roles of small RNA pathways in reproductive medicine Mol Endocrinol 2011. [Epub ahead of print], 160Wang X, Tang S, Le SY, et al. Aberrant expression of oncogenic and tumor-suppressive microRNAs in cervical cancer is required for cancer cell growth PLoS One 2008; 3: e2557.]. Although HPV does not encode miRNAs, it has been shown to affect host miRNA which could provide alternative avenues for the development of nucleic acid-based therapeutics. For instance, the viral E6 protein has been shown to promote cellular proliferation by reducing the expression of the tumor suppressor miR-34a [161Wang X, Wang HK, McCoy JP, et al. Oncogenic HPV infection interrupts the expression of tumor-suppressive miR-34a through viral oncoprotein E6 RNA 2009; 15: 637-47.] and to promote cell migration by downregulation of miR-23b, a p53 target gene [162Au Yeung CL, Tsang TY, Yau PL, Kwok TT. Human papillomavirus type 16 E6 induces cervical cancer cell migration through the p53/microRNA-23b/urokinase-type plasminogen activator pathway Oncogene 2011; 30: 1-10.]. Additionally, the viral E7 protein targets miR-203 to prevent the downregulation of the p63 family of transcription factors in order to promote viral genome amplification in differentiated keratinocytes [163Melar-New M, Laimins LA. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins J Virol 2010; 84: 5212-21.]. However, several issues such as stability, delivery, and off-target effects of miRNAs remain to be addressed to realize their potential as antiviral agents.
For most of the existing HPV inhibitors, the weak antiviral effects observed in cellular assays suggest that further improvements in potency and cell penetration are required. However, it should be noted that all of the HPV inhibitors described in this review have been identified using biochemical assays or surrogate cell-based assays, such that their true antiviral activity is not known. While the use of in vitro assays facilitates the identification of active HPV compounds, it would be of great importance to determine their efficacy in more biologically relevant cellular or animal models of viral pathogenesis. For instance, the use of organotypic raft cultures of keratinocytes immortalized with high-risk HPV genomes is capable of reproducing the complete HPV life cycle leading to virion production, thereby allowing for a detailed mechanistic study of all stages of the viral life cycle [164Meyers C, Mayer TJ, Ozbun MA. Synthesis of infectious human papillomavirus type 18 in differentiating epithelium transfected with viral DNA J Virol 1997; 71: 7381-6.-167Ozbun MA. Infectious human papillomavirus type 31b: purification and infection of an immortalized human keratinocyte cell line J Gen Virol 2002; 83: 2753-63.]. Although there are no definitive animal models that are predictive of the efficacy of HPV antiviral inhibitors in humans, animal models of HPV carcinogenesis exist [168Campo MS. Animal models of papillomavirus pathogenesis Virus Res 2002; 89: 249-61.]. E6/E7 transgenic mice, for instance, can be used to evaluate inhibitors that affect the functions of high-risk E6 and E7, with the caveat that another ligase other than E6AP may be involved in mediating some of the activities of E6 in mice [134Massimi P, Shai A, Lambert P, Banks L. HPV E6 degradation of p53 and PDZ containing substrates in an E6AP null background Oncogene 2008; 27: 1800-4., 169Shai A, Pitot HC, Lambert PF. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice Cancer Res 2010; 70: 5064-73.]. The CRPV infection model, which has been used extensively for pre-clinical studies of vaccine efficacy [170Brandsma JL, Shlyankevich M, Zelterman D, Su Y. Therapeutic vaccination of rabbits with a ubiquitin-fused papillomavirus E1 E2 E6 and E7 DNA vaccine Vaccine 2007; 25: 6158-3.-172Mejia AF, Culp TD, Cladel NM, et al. Preclinical model to test human papillomavirus virus (HPV) capsid vaccines in vivo using infectious HPV/cottontail rabbit papillomavirus chimeric papillomavirus particles J Virol 2006; 80: 12393-7.], could also be used provided that the compounds are active against the CRPV proteins. Although demonstrating activity in an animal model of HPV pathogenesis would be of great value for the development of antivirals, it should not be regarded as essential given the limitations of existing models in mimicking the disease in humans.
The success of these inhibitors will also greatly depend on their ability to remain efficacious against most, if not all, of the prevalent HPV types, with the ultimate hope that a pan-HPV inhibitor can be identified. It is also anticipated that basic research on the HPV life cycle will lead to the identification and validation of novel viral-host proteins interactions that can be exploited for the discovery of inhibitors capable of blocking critical stages of the viral life cycle. As the HPV genome is replicated by cellular DNA polymerases with high fidelity, the emergence of drug-resistant viruses is not expected to be an issue. Thus, treatment with a single antiviral agent (i.e., monotherapy) should, in principle, be sufficient. However, in a clinical setting, it may be beneficial to simultaneously target different steps of the viral life cycle or to combine an antiviral compound with an immuno-stimulatory approach to achieve a more effective treatment of HPV infection. It is hoped that the studies presented above will provide an impetus for the development of better inhibitors and ultimately lead to the discovery of the first HPV antiviral drug capable of alleviating the burden of HPV infections, notably cervical cancer.
ABBREVIATIONS
ACKNOWLEDGEMENTS
We thank Dr. Paola Di Lello for help with the structural representations. Work in the authors’ laboratory is supported by grants from the Canadian Institutes of Health Research (CIHR), Canadian Cancer Society Research Institute, and The Cancer Research Society Inc. C.M.D holds a fellowship from the Fonds de la Recherche en Santé du Québec (FRSQ).
CONFLICT OF INTEREST
None to declare.