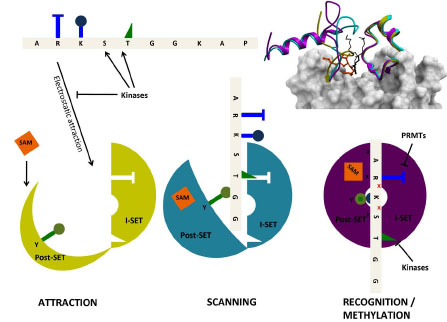
Fig. (5) Structural mechanism of peptide recognition. Observations from the apo (yellow, PDB code 1H3I), binary (cyan, PDB code
1N6C), and ternary (magenta, PDB code 1O9S) structures of SETD7 (top right) can be integrated in a general model for peptide recognition.
Long-range electrostatics attract non-specifically a loose, negatively charged binding groove (where the I-SET domain is already well structured,
but the Pos-SET not) to positively charged histone tails (bottom left). The I-SET domain acts as a rigid reading platform that scans the
histone sequence. SAM binding brings the Post-SET domain to a partially folded state, but the binding groove remains sufficiently open to
allow peptide motion (center). Once a specific sequence is recognized by the I-SET domain, the Post-SET can close in a catalytically competent
conformation (right), the catalytic tyrosine (green) projects towards the site of methyl transfer, and a conserved double hydrogen-bond is
engaged with I-SET (red crosses). Histone marks deposited by other enzymes, such as kinases or protein arginine methyltransferases
(PRMTs), on residues flanking the substrate lysine, can antagonize electrostatic attraction (left) and sequence-specific recognition (right).