- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Inhibition of Inducible Nitric Oxide Synthase Expression by a Novel Small Molecule Activator of the Unfolded Protein Response
Kent T Symons1, Mark E Massari1, Sara J Dozier1, Phan M Nguyen1, David Jenkins1, Mark Herbert2, Timothy C Gahman2, Stewart A Noble2, Natasha Rozenkrants3, Yan Zhang3, Tadimeti S Rao3, Andrew K Shiau1, Christian A Hassig*, 1, 4
Abstract
The transcription of inducible nitric oxide synthase (iNOS) is activated by a network of proinflammatory signaling pathways. Here we describe the identification of a small molecule that downregulates the expression of iNOS mRNA and protein in cytokine-activated cells and suppresses nitric oxide production in vivo. Mechanistic analysis suggests that this small molecule, erstressin, also activates the unfolded protein response (UPR), a signaling pathway triggered by endoplasmic reticulum stress. Erstressin induces rapid phosphorylation of eIF2α and the alternative splicing of XBP-1, hallmark initiating events of the UPR. Further, erstressin activates the transcription of multiple genes involved in the UPR. These data suggest an inverse relationship between UPR activation and iNOS mRNA and protein expression under proinflammatory conditions.
Article Information
Identifiers and Pagination:
Year: 2008Volume: 2
First Page: 1
Last Page: 9
Publisher Id: CCGTM-2-1
DOI: 10.2174/1875397300802010001
Article History:
Received Date: 11/6/2008Revision Received Date: 15/8/2008
Acceptance Date: 20/8/2008
Electronic publication date: 27/9/2008
Collection year: 2008
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Department of Molecular Biology, The Scripps Research Institute, 10550 N. Torrey Pines Road, La Jolla, CA 92037, USA; E-mail: chassig@kalypsys.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 11-6-2008 |
Original Manuscript | Inhibition of Inducible Nitric Oxide Synthase Expression by a Novel Small Molecule Activator of the Unfolded Protein Response | |
INTRODUCTION
Nitric oxide (NO) is an important biological second messenger that regulates a wide array of physiologic processes, including vasodilation, neurotransmission and immune responses. NO is generated by three enzymes; the endothelial, neuronal, and inducible nitric oxide synthases (NOSs). Expression of the inducible NOS (iNOS) gene is upregulated in various cell types in response to proinflammatory agents. As a result, in certain pathophysiological conditions, such as arthritis, asthma and inflammatory bowel disease, the NO produced by iNOS contributes to local inflammation, tissue damage, and pain [1Vallance P, Leiper J. Blocking NO synthesis: how, where and why? Nat Rev Drug Discov 2002; 1: 939-50.]. Transcriptional and post-transcriptional regulation of iNOS is governed by a number of well characterized signaling pathways [2Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase Eur J Pharmacol 2004; 500: 255-66.], however, additional undiscovered pathways of regulation may exist.
The endoplasmic reticulum (ER) is an intracellular organelle responsible for the proper synthesis and folding of secreted and membrane-bound proteins. Many disturbances, including aberrant calcium regulation, altered redox status, or interference with protein glycosylation, can result in the accumulation of unfolded ER client proteins in a phenomenon referred to as ER stress. To compensate for this increased protein burden, a signaling pathway known as the unfolded protein response (UPR) is activated [3Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the Mammalian unfolded protein response Annu Rev Cell Dev Biol 2002; 18: 575-99.]. We have identified a novel small molecule activator of the unfolded protein response (UPR) pathway in a chemical genetic screen for inhibitors of iNOS. The data are consistent with the hypothesis that activation of the UPR leads to specific downregulation of cytokine-induced iNOS production.
MATERIALS AND METHODOLOGY
Cell Culture
A172 human glioblastoma cells, RAW 264.7 mouse macrophage cells, HEK293 and HeLa human adenocarcinoma cells (ATCC) were cultured in DMEM containing 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin. A172 cells were stimulated to express iNOS by adding a cocktail containing hIFN-γ (4,000 U/ml), hTNF-α (40 ng/ml), and hIL-1β (4 ng/ml). RAW 264.7 cells were stimulated with lipopolysacharride (1 µg/ml) (Sigma) and mIFN-γ (100 U/ml) (Roche Diagnostics) unless otherwise indicated.
Nitric Oxide Detection
A172 or RAW 264.7 cells were plated in 384- or 1536-well plates and simultaneously treated with stimulation media and compound. After 18 hours of incubation NO production was measured by adding 2,3-diaminonaphthalene (DAN) (10 μg/ml final) (Invitrogen) diluted in DMEM supplemented with hydrochloric acid (0.1N final) and incubated for 20 minutes at room temperature. Fluorescence was measured after increasing pH with sodium hydroxide (0.12N final) [4Misko TP, Schilling RJ, Salvemini D, Moore WM, Currie MG. A fluorometric assay for the measurement of nitrite in biological samples Anal Biochem 1993; 214: 11-6.].
Cytoblot
Protocol was modified from a previously described assay [5Stockwell BR, Haggarty SJ, Schreiber SL. High-throughput screening of small molecules in miniaturized mammalian cell-based assays involving post-translational modifications Chem Biol 1999; 6: 71-83.]. Briefly, RAW 264.7 cells were plated in 384-well format and simultaneously treated with stimulation media and compound. After 18 hours incubation, cells were fixed with ethanol / acetic acid (95% / 5%) and blocked with PBS/ 4% BSA for 1 hour at room temperature. Cells were incubated with a 1:250 dilution of iNOS primary antibody (BD Biosciences) in PBS/ 1% BSA for 1.5 hrs at room temperature. A 1:2500 dilution of donkey anti-mouse HRP conjugated secondary antibody (Jackson ImmunoSciences) in PBS/ 1% BSA was added to wells and incubated 1 hour at room temperature. HRP activity was detected with luminol (Santa Cruz Biotech) on an Analyst GT plate reader.
Western Blots
RAW 264.7 cells were incubated with stimulation media and compound at the indicated times and concentrations.
Whole cell extracts were prepared in the presence of protease inhibitors (Roche) and for eIF-2α, phosphatase inhibitors were added (Pierce). Total protein concentration was measured using the Advanced Protein Assay (Cytoskeleton, Inc) and samples were normalized for loading. Samples were run on 4-20% tris-glycine gels or 4-12% Bis-Tris gels (Invitrogen) and electro-transferred (Biorad) to nitrocellulose membranes (Invitrogen). For iNOS detection, membranes were blocked overnight in tris-buffered saline containing 0.05% (v/v) Tween-20 (TBST) and 3% (w/v) nonfat milk. Membranes were then incubated with a 1:2500 dilution of murine iNOS (BD Biosciences) primary antibody for 1 hour. This was followed by incubation with 1:2000 dilution of HRP conjugated goat anti-mouse secondary for 1 hour. For eIF2α and eIF2α phosphoSer51, membranes were blocked over night in Starting Block (Pierce) containing 0.1% (v/v) Tween-20 and incubated in a 1:500 dilution of either anti-eIF-2α or anti-eIF-2α-phospho S51 (Cell Signalling Technologies) for 2 hours, followed by incubation with 1:2000 dilution of HRP-conjugated secondary antibody for 1 hour. For phospho-STAT1 (Tyr 701) and phospho-STAT3 (Tyr 705), membranes were blocked overnight in TBST with 5% (w/v) nonfat milk. Blots were incubated with 1:1000 dilution of the appropriate STAT antibody (Cell Signaling Technology) for 2 hours, followed by incubation with goat anti rabbit HRP (1:2000) for 1 hour. All membranes were developed with WestDura substrate (Pierce) and imaged using a Fluor-Chem imager (Alpha Innotech Corporation).
XBP-1 Splicing Assay
HeLa cells were seeded in 6-well dishes and treated with compound for 1 hour and washed twice with PBS. RNA was isolated using the RNeasy kit including on-column DNase treatment (Qiagen). cDNA was transcribed using SuperscriptIII (Invitrogen) and PCR of a 597bp nucleotide fragment, including the 26 nucleotide alternately spliced intron, was performed as described previously [6DuRose JB, Tam AB, Niwa M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress Mol Biol Cell 2006; 17: 3095-107.].
Quantitative Real Time PCR and Microarray Analysis
After incubation with compound and stimulation media, A172 or RAW 264.7 cells were lysed in 350 μl buffer RLT, homogenized over QiaShredder columns, and total RNA was purified using the RNeasy kit with on-column DNase digestion (Qiagen). RNA quality and concentration was determined using the Bioanalyzer RNA6000 assay (Agilent). 2 µg of RNA was used in cDNA synthesis reactions (Invitrogen). Each cDNA sample was amplified using SYBRgreen-based qPCR and a melting curve was determined. For microarray studies, 5 µg of the RNA was analyzed on U133A 2.0 Gene Chips using standard protocols (Affymetrix). Analysis was conducted using gcRMA normalization with GeneSpring and Spotfire software.
Cytotoxicity
A172 cells were plated in 1536-well plates at 2,500 cells/well and allowed to adhere for 4 hours. Subsequently the cells were simultaneously stimulated with cytokine and treated with compound. Cells were then incubated for 6 or 18 hours followed by addition of ATPlite (Perkin Elmer) to determine cellular ATP levels. Luminescence was read on an Analyst GT microplate reader. For mitochondrial membrane potential measurements, stimulated A172 human glioblastoma cells were treated with 10 µM ToxoFlavin, 10 µM erstressin or 10 µM nostressin in DMEM with 0.1% DMSO, 5% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin for 6 or 18 hours. Cells were trypsinized and washed twice with PBS. Cells were resuspended in 100 µl staining buffer containing 5 µg/ml JC-1 dye (Invitrogen). After incubating for 15 minutes at 37ºC, cells were acquired for fluorescent measurement using a Becton Dickinson LSRII flow cytometer. After gating on live cells, those which exhibited a fluorescent shift from 590nm to 525nm were considered apoptotic [7Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye Methods Enzymol 1995; 260: 406-17.].
Rat LPS Model
Male Lewis rats (Charles River) 150-200 g were administered a dose of 0.3 mg/kg of lipopolysaccharride - LPS (Sigma) dissolved in 0.9% sodium chloride. The intravenous injection was given though the penal vein under anesthesia with 5% isoflurane in medical grade pure oxygen and maintained at 2-3% isoflurane. Drug (30 mg/kg in Encapsin, 5% DMSO) or vehicle alone was administered 3 to 5 minutes prior to LPS challenge via intraperitoneal injection. Rats were returned to their home cages and sacrificed 6 hours later. Blood samples were collected for analysis of nitrates (Cayman Chemical Nitrate/Nitrite Flourometric Assay Kit). Fluorescence was measured using an Aquest plate reader (Molecular Devices). All results are expressed as mean + SEM.
NFκB Electrophoretic Mobility Shift Assays
DNA binding assays were performed as described previously. Briefly, 10 μg of nuclear extract was mixed with 1 μl of 10 mg/ml BSA, 0.5 μl of 1 mg/ml poly (dI:dC), 50,000 cpm of 32P-end-labeled probe, 2 μl of 10X binding buffer (100 mM Tris Cl, pH 7.5, 500 mM NaCl, 10 mM DTT, 10 mM EDTA and 50% glycerol) and 1 μl of antiserum (where appropriate) in a 20 μl final reaction volume. Following 15 minutes of incubation at room temperature, complexes were resolved on a 5% native polyacrylamide gel in 0.5× TBE (45 mM Tris, 45 mM boric acid, 1 mM EDTA). The oligonucleotide probe (top strand) used in this study was NFκB, 5’-AGT TGA GGG GAC TTT CCC AGG C-3’. NFκB antibody was purchased from Santa Cruz Biotechnologies [8Bain G, Gruenwald S, Murre C. E2A and E2-2 are subunits of B-cell-specific E2-box DNA-binding proteins Mol Cell Biol 1993; 13: 3522-9.,9Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei Nucleic Acids Res 1983; 11: 1475-89.].
RESULTS AND DISCUSSION
To identify small molecules that reduce the production of iNOS-derived NO, we developed a homogeneous forward chemical genetic screen in the murine macrophage cell line RAW 264.7. Stimulation of RAW 264.7 cells with bacterial lipopolysaccharide (LPS) and IFN-γ results in the increased expression of a host of inflammatory genes including iNOS [10Kleinert H, Schwarz PM, Forstermann U. Regulation of the expression of inducible nitric oxide synthase Biol Chem 2003; 384: 1343-64.]. The effect of compounds on iNOS activity was quantified by indirectly measuring the production of NO from cells. This approach enables not only the detection of compounds that inhibit iNOS directly, but also compounds that act upstream in the iNOS-NO axis. Using this assay, we screened a 650,000 compound library using a fully automated ultra high-throughput robotic system. Compounds were tested in single point at 10 μM and hits were confirmed in 7-point dose response. A total of 330 compounds (0.05% hit rate) demonstrated significant reduction in NO production (>30% inhibition) without detectable cytotoxicity. One inhibitor identified from this screen, compound 1, shared no structural similarity to previously described iNOS inhibitors (Fig. 1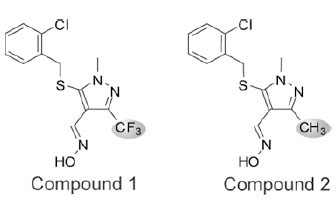 ). Several analogs of compound 1 were synthesized and tested in the homogeneous cell-based NO detection and other follow-up assays. These analogs provided a cursory assessment of the structure activity relationship for the series. Replacement of the R1 2-chlorobenzylthio with the 3-methyl derivative (Cmpd 3) was tolerated, though removal of the 2-substituent (Cmpd 4) or addition of a 6-fluoro substituent (Cmpd 5) reduced activity across all assays. Modifications to the R2 oxime through alkylation (Cmpd 6), conversion to a hydrazone (Cmpds 7 and 8) or replacement with a carboxylic acid or amide (Cmpds 9 and 10) greatly reduced or eliminated activity. Two additional structurally related analogs of merit were identified (Cmpds 11 and 12) with R1 sulfones in place of the thio ether and a nitrile in place of the R2 oxime. Compound 12 displayed the most potent in vitro activity profile of the series, and the data indicate that the oxime is not essential for activity of compounds within this chemical series. Strikingly, replacement of the R3 trifluoromethyl with a methyl group (Cmpd 2) resulted in an inactive molecule (Fig. 1 and Table 1). In contrast to compound 1, which inhibited NO production with an IC50 of 2.8 μM, compound 2 was essentially inactive in the NO detection assay (Fig. 2A
). Several analogs of compound 1 were synthesized and tested in the homogeneous cell-based NO detection and other follow-up assays. These analogs provided a cursory assessment of the structure activity relationship for the series. Replacement of the R1 2-chlorobenzylthio with the 3-methyl derivative (Cmpd 3) was tolerated, though removal of the 2-substituent (Cmpd 4) or addition of a 6-fluoro substituent (Cmpd 5) reduced activity across all assays. Modifications to the R2 oxime through alkylation (Cmpd 6), conversion to a hydrazone (Cmpds 7 and 8) or replacement with a carboxylic acid or amide (Cmpds 9 and 10) greatly reduced or eliminated activity. Two additional structurally related analogs of merit were identified (Cmpds 11 and 12) with R1 sulfones in place of the thio ether and a nitrile in place of the R2 oxime. Compound 12 displayed the most potent in vitro activity profile of the series, and the data indicate that the oxime is not essential for activity of compounds within this chemical series. Strikingly, replacement of the R3 trifluoromethyl with a methyl group (Cmpd 2) resulted in an inactive molecule (Fig. 1 and Table 1). In contrast to compound 1, which inhibited NO production with an IC50 of 2.8 μM, compound 2 was essentially inactive in the NO detection assay (Fig. 2A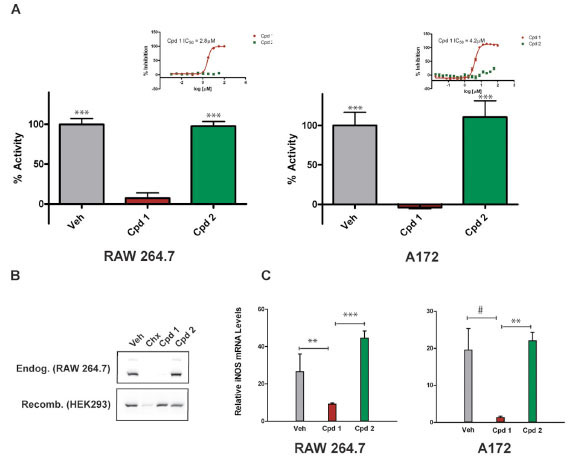 ). Demonstrating that its effects are conserved across species, compound 1 also inhibited NO production in cytokine-stimulated human A172 glioblastoma cells with an IC50 of 4.2 μM. As seen in RAW264.7 cells, compound 2 did not affect NO production in A172 cells (Fig. 2A). The subtle structural difference between compound 1 and compound 2, combined with the significant difference in ability of the two compounds to block cellular iNOS activity, facilitated a pharmacological approach to elucidating the mechanism of action of compound 1.
). Demonstrating that its effects are conserved across species, compound 1 also inhibited NO production in cytokine-stimulated human A172 glioblastoma cells with an IC50 of 4.2 μM. As seen in RAW264.7 cells, compound 2 did not affect NO production in A172 cells (Fig. 2A). The subtle structural difference between compound 1 and compound 2, combined with the significant difference in ability of the two compounds to block cellular iNOS activity, facilitated a pharmacological approach to elucidating the mechanism of action of compound 1.


Interestingly, a counter screen for biochemical enzyme inhibition revealed that compound 1 failed to reduce iNOS activity, indicating that the compound might not directly inhibit the enzyme (data not shown). A tertiary 384-well microplate-based in-cell western blot assay (cytoblot) in RAW264.7 cells utilizing an iNOS-specific antibody determined that compound 1 inhibited LPS/IFN-γ-induced iNOS protein expression with an IC50 of 1.5 μM, a value comparable to that calculated from the NO detection assay (Supplemental Fig.
F1). To confirm the cytoblot findings, we performed immunoblot analysis and demonstrated that 10 μM compound 1 blocked iNOS expression in RAW264.7 cells. This effect was not seen when cells were treated with vehicle or compound 2, but was seen when global protein translation was blocked in cells using cycloheximide (Fig. 2B, top panel). In contrast to the effects of compound 1 on endogenous iNOS, heterologous expression of recombinant iNOS in HEK293 cells was unperturbed by either compound 1 or compound 2, whereas expression was reduced in cells treated with cycloheximide (Fig. 2B, bottom panel). These data indicate that compound 1 acts upstream in the iNOS pathway and reduces NO production in cells by reducing iNOS protein levels.
Next, we tested the effects of compound on cellular iNOS mRNA transcript levels. A172 cells and RAW264.7 cells were stimulated with proinflammatory factors and treated with compounds at a final concentration of 10 μM for 6 hours before isolating the RNA and performing real-time quantitative PCR to determine whether iNOS mRNA levels were affected. As seen in Fig. (2C), a statistically significant reduction in iNOS transcripts was measured following treatment with compound 1 relative to either compound 2 or vehicle (DMSO). The reduction of iNOS mRNA caused by compound 1 relative to compound 2 ranged from approximately 4.7-fold in RAW264.7 macrophages (p = 0.01) to 13.7-fold in A172 cells (p = 0.045). As seen for iNOS protein, compound 2 had no statistical effect on iNOS mRNA levels relative to vehicle (Fig.
F2C). These results indicate that compound 1 reduces NO production in cytokine-stimulated cells, at least in part, by reducing cellular iNOS mRNA levels.
To identify the mechanism whereby compound 1 inhibited iNOS expression and to identify the biological pathway(s) impacted by compound 1, we then profiled global gene expression in A172 cells simultaneously treated with cytokine-stimulation media and compound. A significant difference was defined as ≥ 2-fold change (up or down) in mRNA transcript levels with a p value ≤0.05. In cells stimulated with cytokines for 30 minutes, no significant differences in gene expression were observed between cells treated with compound 1 or compound 2 at 10 μM. However, as compared to the 30 minute time-point, after 6 hours of incubation with proinflammatory cytokines and vehicle, approximately 3.8% of genes profiled exhibited altered expression, consistent with the well documented activation of inflammatory signaling pathways [11Hume DA, Wells CA, Ravasi T. Transcriptional regulatory networks in macrophages Novartis Found Symp 2007; 281: 2-18.]. Strikingly, at 6 hours there was virtually no difference in the gene expression profile (< 0.1 % of transcripts) of cells treated with compound 2 and cytokine as compared to cytokine treatment alone. In contrast, cytokine stimulated cells treated with compound 1 displayed a significantly different gene expression profile at the 6 hour time-point. A subset of genes with altered expression in compound 1 treated cells were common to those induced by cytokine stimulation. As anticipated from previously described qPCR results, iNOS mRNA levels, which were induced by cytokine stimulation, were reduced 4.7-fold by coincubation with compound 1 (but unaffected by compound 2 treatment). Notably, in addition to iNOS, the transcripts of several other inflammatory mediators normally upregulated by cytokines, (TNFα, IL1β, MCP-2 and I-TAC) were downregulated by compound 1 relative to compound 2 or vehicle (GEO submission GSE7806). Gene expression was not universally reduced however, for example, transcripts of IL15 (2.4-fold), chemokine orphan receptor 1 (CMKOR1, 11.2-fold) and ATP-binding cassette protein 1 (ABCA1, 3.5-fold) were increased by compound 1 treatment (GEO submission GSE7806).
Interestingly, in addition to altering inflammatory gene expression, compound 1 also affected the mRNA levels of multiple genes involved in the UPR. The transcription factors XBP-1 (2.7-fold), ATF3 (3.2-fold), ATF4 (3.3-fold), and CHOP (22-fold) were significantly induced in compound 1-treated cells. Molecular chaperones ERdj4 (24-fold) and BiP (2.7-fold) and ER to Golgi transporters SEC23B (2-fold) and SEC31L1 (4-fold) were also significantly elevated by compound 1. Further, Herpud1/ Herp, a membrane-associated protein which functions in ER associated degradation was upregulated 9.7-fold (Fig. 3A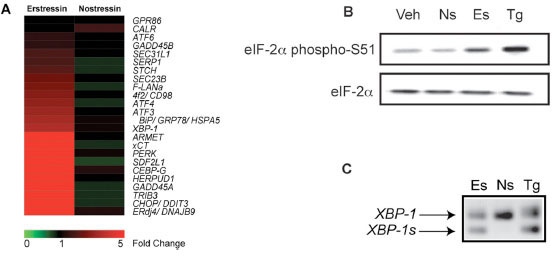 ). Changes in the expression of several genes identified in the expression profiling study were confirmed by independent qPCR analysis (Supplemental Figs.
F2A and 2B). Similar gene expression signatures have been described for known ER stressors, including tunicamycin and thapsigargin [12Donati G, Imbriano C, Mantovani R. Dynamic recruitment of transcription factors and epigenetic changes on the ER stress response gene promoters Nucleic Acids Res 2006; 34: 3116-27., 13Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response Mol Cell Biol 2003; 23: 7448-59.] and these effects were absent in cells treated with compound 2. These and other findings described below suggest that compound 1 may induce the UPR, and as a result, we have named this compound erstressin and its inactive analog compound 2, nostressin.
). Changes in the expression of several genes identified in the expression profiling study were confirmed by independent qPCR analysis (Supplemental Figs.
F2A and 2B). Similar gene expression signatures have been described for known ER stressors, including tunicamycin and thapsigargin [12Donati G, Imbriano C, Mantovani R. Dynamic recruitment of transcription factors and epigenetic changes on the ER stress response gene promoters Nucleic Acids Res 2006; 34: 3116-27., 13Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response Mol Cell Biol 2003; 23: 7448-59.] and these effects were absent in cells treated with compound 2. These and other findings described below suggest that compound 1 may induce the UPR, and as a result, we have named this compound erstressin and its inactive analog compound 2, nostressin.

Activation of the UPR gene expression program is initiated by at least three ER transmembrane protein sensors, including ATF-6, IRE-1, and PERK. Under normal conditions, all three proteins are maintained in inactive conformations by Grp78, an HSP70-family chaperone. The binding of accumulated unfolded proteins to Grp78 causes the dissociation of these three complexes and the activation of signal transduction. IRE-1 and PERK are both kinases that self-oligomerize upon release of Grp78. In the case of IRE-1, oligomerization-induced autophosphorylation activates an internal ribonuclease domain that triggers alternative splicing of its mRNA substrate, XBP-1, encoding a bZIP transcription factor that activates downstream UPR genes. Oligomerized PERK undergoes autophosphorylation and then phosphorylates two substrates, the bZIP transcription factor NRF2 and elongation factor 2α (eIF2α). Cumulatively, these signaling events lead to the UPR gene transcription response [14Schroder M, Kaufman RJ. Divergent roles of IRE1alpha and PERK in the unfolded protein response Curr Mol Med 2006; 6: 5-36.].
To determine if erstressin affects UPR gene expression by activating these pathways, we tested for several early markers of ER stress signaling. For comparison, we also characterized thapsigargin, which induces ER stress by inhibiting the Ca2+-ATPase resident within the ER, thereby causing protein misfolding indirectly by disrupting the function of Ca2+-dependent chaperones Grp78, Grp94, and calreticulin [15Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases Trends Pharmacol Sci 1998; 19: 131-5.]. Immunoblot analysis of protein extracts isolated from RAW264.7 cells treated with 10 μM erstressin for 30 minutes showed elevated eIF2α phosphorylation. This effect by erstressin was less pronounced than that seen in cells treated with 1 μM thapsigargin, but was completely absent in cells treated with 10 μM nostressin (Fig. 3B). Next, we tested whether the splicing of XBP-1 mRNA by the ER transmembrane ribonuclease IRE-1, was altered by erstressin treatment. PCR amplification of RNA samples isolated from HeLa cells incubated with 20 μM erstressin or 50 nM thapsigargin for 1 hour revealed the presence of alternatively spliced XBP-1. Once again, 20 μM nostressin did not induce alternative splicing of XBP-1 (Fig. 3C). These findings suggest that, similar to thapsigargin, erstressin activates multiple UPR signaling pathways within the first 0.5 - 1 hour of compound treatment. The eIF2α phosphorylation was increased by erstressin treatment prior to any detectable changes in transcription (within 30 minutes), suggesting that this effect by erstressin is upstream of changes in UPR gene expression.
To eliminate the possibility that the effects of erstressin on iNOS were simply a consequence of reduced cell viability or general cytotoxicity, we tested the effects of the compound on cytokine induced A172 cells in several assays. Reduced metabolic activity and cell viability as a consequence of the depletion of cellular ATP stores was measured in cells treated with compound. Cellular ATP levels were unaffected at both 6 hours and 18 hours of treatment with either erstressin or nostressin, but substantially reduced in cells treated with the positive control, toxoflavin (Fig. 4A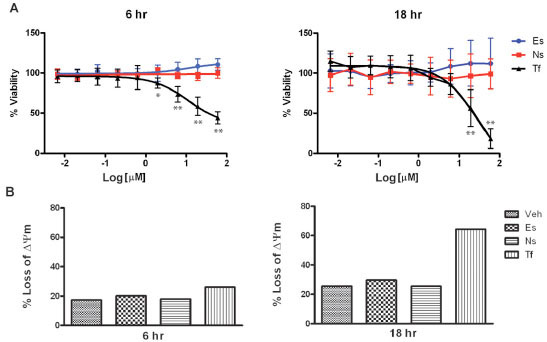 ). In separate experiments, cells were stained with JC-1, a cationic dye which accumulates in the mitochondria under depolarized conditions, and can be used to detect reduced viability and apoptosis in individual cells. As anticipated from the ATP assay, no increase in JC-1-positive cells was observed at 6 hours or 18 hours of treatment with either erstressin or nostressin, even at concentrations of 10 μM or 30 μM (Fig.4B and data not shown). Similar results were obtained using an independent apoptosis assay (Annexin V) (data not shown). These findings suggest that cytotoxicity is unlikely to account for the observed reduction in iNOS expression and activity caused by erstressin in A172 cells.
). In separate experiments, cells were stained with JC-1, a cationic dye which accumulates in the mitochondria under depolarized conditions, and can be used to detect reduced viability and apoptosis in individual cells. As anticipated from the ATP assay, no increase in JC-1-positive cells was observed at 6 hours or 18 hours of treatment with either erstressin or nostressin, even at concentrations of 10 μM or 30 μM (Fig.4B and data not shown). Similar results were obtained using an independent apoptosis assay (Annexin V) (data not shown). These findings suggest that cytotoxicity is unlikely to account for the observed reduction in iNOS expression and activity caused by erstressin in A172 cells.

To evaluate the potential of erstressin to suppress iNOS expression in vivo, we determined the effects of erstressin on NO production in a model of inflammatory disease. Administration of LPS to rodents induces a systemic inflammatory response coincident with expression of iNOS [16Kato H, Negoro M, Wakabayashi I. Effects of acute ethanol administration on LPS-induced expression of cyclooxygenase-2 and inducible nitric oxide synthase in rat alveolar macrophages Alcohol Clin Exp Res 2005; 29: 285S-93S.]. Consistent with its ability to reduce iNOS activity in cell culture, intraperitoneal administration of erstressin reduced NO metabolites in rat plasma following LPS stimulation by 50% relative to vehicle, p<0.001 (Fig. 5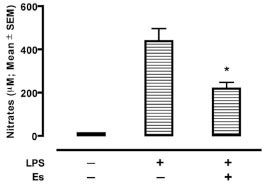 ). In a separate experiment, the potent iNOS inhibitor, 1400W, was tested as a comparator in the LPS model (Supplemental Fig.
3). Although 1400W is a more efficacious compound (76% reduction in NO metabolites; p<0.0001), the results are consistent with an ability of erstressin to downregulate iNOS in vivo during proinflammatory stimulation, providing for the possibility that a similar mechanism of action to that seen in cell culture experiments might downregulate iNOS in certain inflammatory disease settings.
). In a separate experiment, the potent iNOS inhibitor, 1400W, was tested as a comparator in the LPS model (Supplemental Fig.
3). Although 1400W is a more efficacious compound (76% reduction in NO metabolites; p<0.0001), the results are consistent with an ability of erstressin to downregulate iNOS in vivo during proinflammatory stimulation, providing for the possibility that a similar mechanism of action to that seen in cell culture experiments might downregulate iNOS in certain inflammatory disease settings.

CONCLUSION
We have conducted a series of chemical genetic studies on iNOS expression that has led to the identification and initial characterization of erstressin, a unique methylpyrazole-based inducer of the UPR. The chemical structure of erstressin is novel and does not immediately reveal clues as to the identity of its molecular target(s). Initial structure activity relationship studies indicate that the substitution and derivitization of the scaffold might enable synthesis of an affinity matrix for isolation of cellular targets (TG, CH unpublished observations). The synthetic tractability of the series should help facilitate the identification of its receptor and provide a useful tool to further investigate upstream components of the UPR pathway.
A direct relationship between the UPR-inducing properties of erstressin and the ability of the compound to block iNOS expression can be inferred from several findings. First, the ability of erstressin to downregulate iNOS expression is inextricably linked to its ER stressor activity, as a subtle change in chemical structure (nostressin) abrogates the effects in both pathways. The concentrations of erstressin required to activate UPR signaling and UPR gene expression are the same as those that reduce iNOS mRNA and protein expression and enzyme activity. The structurally unrelated ER stressor, thapsigargin, also reduced iNOS activity, protein levels, and mRNA transcripts (data not shown). However, thapsigargin is somewhat cytotoxic at efficacious concentrations, making it difficult to separate a specific effect on iNOS from general cytotoxicity. Although CHOP (an apoptosis-associated gene correlated with the UPR) expression is elevated in response to erstressin treatment in A172 cells, the effects on iNOS expression levels or activity are not a consequence of reduced viability or programmed cell death induced by the compound. Importantly, erstressin treatment results in specific gene expression changes and does not appear to cause a global block in transcription. Erstressin does not appear to act by interfering with STAT phosphorylation (Supplemental Fig. 4A) or NFκB DNA binding (Supplemental Fig. 4B), two key transcriptional activators of iNOS. Thus, we propose that erstressin might block iNOS transcription by inducing ER stress and/or activating the UPR by an as yet unknown mechanism.
A negative regulatory role for ER stress in iNOS expression has been proposed recently for several known and putative UPR inducers [17Takano Y, Hiramatsu N, Okamura M, et al. Suppression of cytokine response by GATA inhibitor K-7174 via unfolded protein response Biochem Biophys Res Commun 2007; 360: 470-5.]. Interestingly, a similar effect on iNOS has been described for resveratrol and its analogs, and we speculate that UPR activation might contribute to the anti-inflammatory activity of these compounds [18Park JW, Woo KJ, Lee JT, et al. Resveratrol induces pro-apoptotic endoplasmic reticulum stress in human colon cancer cells Oncol Rep 2007; 18: 1269-73.-21Meng XL, Yang JY, Chen GL, et al. RV09, a novel resveratrol analogue, inhibits NO and TNF-alpha production by LPS-activated microglia Int Immunopharmacol 2008; 8: 1074-82.]. To a large extent however, previous studies have suggested a general proinflammatory role for ER stress [22Zhang K, Shen X, Wu J, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response Cell 2006; 124: 587-99.]. While the precise molecular details whereby activation of the UPR results in transcriptional downregulation of iNOS have yet to be uncovered, we propose that both pathways could compete for common transcription factors or coactivators, resulting in the observed transcriptional attenuation. Consistent with this idea, NFκB, ATF-3, and p300/CBP have been suggested to activate transcription of both inflammatory, as well as UPR genes [12Donati G, Imbriano C, Mantovani R. Dynamic recruitment of transcription factors and epigenetic changes on the ER stress response gene promoters Nucleic Acids Res 2006; 34: 3116-27., 22Zhang K, Shen X, Wu J, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response Cell 2006; 124: 587-99.-24Inoue K, Zama T, Kamimoto T, et al. TNFalpha-induced ATF3 expression is bidirectionally regulated by the JNK and ERK pathways in vascular endothelial cells Genes Cells 2004; 9: 59-70.]. Additional mechanisms, such as selective or non-selective translational attenuation, may further contribute to the overall reduction in iNOS protein. Our data indicate that continued investigation of the relationship between these two pathways is warranted and could ultimately lead to new therapies for the treatment of inflammatory disorders involving iNOS.
Supplementary Material
Supplementary material can be viewed at Download Supplementary Material
ACKNOWLEDGEMENTS
Microarray sample preparation, hybridization, and scanning were conducted by the GeneChip Core Laboratory of the Veterans Medical Research Foundation, La Jolla, CA. We thank Dr. Kees Murre and Dr. Maho Niwa for helpful discussion. We are grateful to Ian Wilson and Dennis Wolan for helpful review of the manuscript.
CONFLICT OF INTEREST
All authors hold, or have the option to hold, Kalypsys, Inc. company stock.