
The Open Cell Signaling Journal
(Discontinued)
ISSN: 1876-3901 ― Volume 4, 2012
Scorched Earth or Viral Birth?
Ian Gentle*, 1, 2, Ueli Nachbur*, 1
Abstract
Whether a virus is coded for by RNA, single-stranded DNA or double-stranded DNA (dsDNA), the ultimate goal of the virus is to hijack the host’s biosynthetic machinery to replicate and spread. Like spies fomenting revolt behind enemy lines, viruses have evolved to invade cells and then use stealth and deception to avoid or disrupt the cell’s defences [1]. Cells are not without defences, however, and have evolved a number of ways to detect the presence of viral invaders and prevent their replication. In particular, cells may choose the ‘scorched earth’ policy and kill themselves by activating an apoptotic program to prevent the virus from replicating. Multicellular organisms have evolved to accept a small-scale cellular loss for the greater good, i.e., to prevent the defeat of the entire organism. However, while viruses must evade the cellular apoptosis response in order to replicate and therefore mostly counteract apoptosis, some viruses even manage to subvert this protective response to destroy their hosts at a late stage of infection in order to facilitate release of mature viral particles.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 3
First Page: 35
Last Page: 41
Publisher Id: TOCELLSJ-3-35
DOI: 10.2174/1876390101103010035
Article History:
Received Date: 2/5/2010Revision Received Date: 21/8/2010
Acceptance Date: 22/10/2010
Electronic publication date: 14/4/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to these authors at the La Trobe Institute for Molecular Science, Department of Biochemistry, Bundoora, VIC 3086, Australia; Tel: +61 3 9479 5842; Fax: +61 3 9479 1728; E-mails: Ian.gentle@uniklinik-freiburg.de, U.Nachbur@latrobe.edu.au
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 2-5-2010 |
Original Manuscript | Scorched Earth or Viral Birth? | |
VIRAL DETECTION
Cells use sentinel proteins that recognise pathogen-specific molecules or PAMPs [2] that are located in different cellular compartments. These sentinel proteins are known as pattern recognition receptors (PRRs) and they include the TOLL-like receptors (TLRs), NOD-like receptors (NLRs) and intracellular helicases, typified by Rig-I (retinoic acid-inducible gene I) and sometimes referred to as the Rig-like helicases (RLHs) [3]. The infection cycle of RNA and also some DNA viruses [4, 5] results in double-stranded RNA byproducts that are detected by sentinel proteins such as TLR3, TLR7 and Rig-I [6]. These receptors signal via distinct signalling complexes. For example, TLR7, which is localised in endosomes, binds viral-derived dsRNA that is delivered to the endosomes via an autophagic process [7]. This dsRNA binds and activates the receptor, resulting in NF-κB activation and Type I interferon transcription via the interferon regulatory factors IRF3 and IRF7 (Fig. 1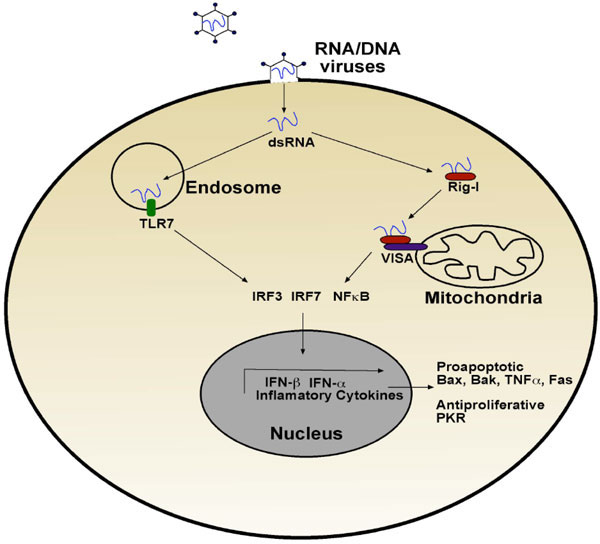 ).
).

Similarly, when dsRNA binds to Rig-I, a cytoplasmic helicase containing two caspase recruitment domains (CARDs), Rig-I translocates to the mitochondria where it interacts via a CARD-CARD interaction with Cardif/Mavs/VISA/IPS-1 [8-10]. Like TLR7, Rig-I activation results in the activation of IRF3/IRF7 and NF-κB, and the production of Type I interferons, interferon α and β and inflammatory cytokines such as tumour necrosis factor alpha (TNFα) (Fig. 1).
Type I interferons are defined by their ability to bind to the interferon α receptor. Type I interferons are secreted by virally infected cells and activate the Jak/STAT pathway which regulates many genes, including a number of pro-apoptotic genes such as Bax, Bak, TRAIL and Fas [11]. In addition, Type I interferon signalling promotes the transcription of proteins with direct antiviral activity. For example, the serine/threonine kinase PKR is induced when viral RNA is detected and subsequently phosphorylates the α subunit of the translation factor eIF2, thereby preventing translation and limiting production of viral proteins. An added effect of loss of translation is that cells become sensitised to death-inducing cytokines such as TNFα, because they are no longer able to mount an anti-apoptotic response. Loss of translation sensitises the cells to TNFα by blocking the NF-κB mediated induction of pro-survival proteins, resulting in the signal routing through the death arm of the TNFα pathway (nicely reviewed by Kantari et al. in this showcase).
Type I interferons also sensitise cells to mitochondria-mediated cell death or intrinsic cell death. One possible mechanism involves the tumour suppressor protein p53, which is a target for interferon α and β [12]. Indeed, p53 can induce the expression of pro-death proteins such as Puma and prevent the transcription of pro-survival proteins such as Bcl-2. Increased p53 levels allow a more potent stress response to viral infection, leading to more rapid apoptosis and lower viral replication.
VIRAL EVASION
Detection of viral products simultaneously puts cells in a hyper-sensitive state where they are poised for death through the interferon response and also upregulates other inflammatory cytokines such as TNFα, alerting the immune system and neighbouring cells of a potential threat. The cell’s ability to sense viral molecules and switch on its early defences is inhibitory to the virus’s successful infiltration, and therefore, viruses have been forced to respond with an impressive arsenal of gadgets to prevent and inhibit their detection. There are a number of tactics that viruses employ to avoid the cell’s innate immune response.
An obvious strategy is to avoid detection altogether. Blocking interferon production is an early point that viruses target to avoid detection and limit the immunological response. The hepatitis C virus protease NS3-4A targets both the TLR-3 adaptor TRIF and the C-terminal region of VISA (virus-induced signalling adapter), thus preventing Rig-I mediated signalling and transcription of interferon α and β (Fig. 2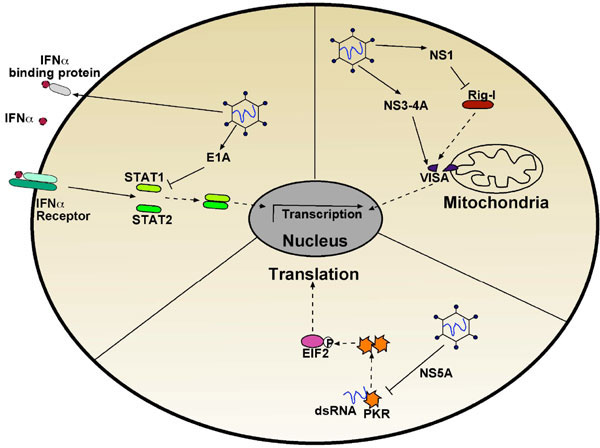 ) [9]. Similarly, the V proteins of paramyxoviruses and NS1 protein of influenza block the RNA helicases mda5 (melanoma differentiation-associated gene 5) and Rig-I, respectively [13].
) [9]. Similarly, the V proteins of paramyxoviruses and NS1 protein of influenza block the RNA helicases mda5 (melanoma differentiation-associated gene 5) and Rig-I, respectively [13].

More downstream events are also tampered with, including signalling from the interferon receptors themselves. Most orthopoxviruses produce soluble interferon α/β binding proteins, which sequester the interferon ligands, preventing their binding to the receptors [14]. Signalling from the receptors may also be inhibited: the V protein of parainfluenza virus 5 targets STAT1 for degradation, and the adenoviral E1A protein interrupts interferon α/β signalling by directly interacting with STAT1 and lowering its levels along with its transcriptional coactivators (Fig. 2) [13]. Despite the virus’s best efforts, interferon signalling may still be activated and many viruses encode proteins to inhibit interferon-induced antiviral proteins such as PKR. For example, the hepatitis C virus protein NS5A can bind to and inhibit PKR directly (Fig. 2) [15].
TLR and RLH pathways share common signalling elements with pro-death and pro-inflammatory cytokines such as TNFα. Inhibition of proteins mediating signalling at both these points is therefore an efficient way for viruses to circumvent the host’s defences. The receptor interacting serine/threonine kinase, RIPK1, mediates signalling from TNF family receptors. Upon ligand binding, RIPK1 is recruited to the receptor via interactions with two other TNF receptor-associated proteins, TRAF2 and TRADD, and becomes ubiquitylated by cIAPs. The IKK kinase complex is activated following its recruitment to K63 polyubiquitylated RIPK1 and also linear ubiquitin chains associated with HOIL and HOIP [16], whereupon it phosphorylates IκB (the NF-κB inhibitor), which is thereby tagged for degradation. RIPK1 is also present in complex II that contains caspase-8 and which is the death-inducing arm of TNFR1. In addition to its role in TNFR1 signalling pathways, RIPK1 is equally important for TLR3 signalling. In this pathway, RIPK1 interacts with TRIF, a kinase regulating TLR3 signalling and in the absence of RIPK1, TLR3-mediated NF-κB activation is abolished [17]. RIPK1 also mediates NF-κB signalling from VISA, the mitochondrial receptor for Rig-I in a manner similar to that of TNFR1 stimulation [18, 19]. RIPK1’s role in multiple NF-κB-mediated defence pathways means it plays a key role in determining a cell’s fate upon viral detection and inflammatory cytokine stimulation, and it is not surprising, therefore, that it is targeted by viruses. Cytomegalovirus protein M45 was shown to bind to and inhibit RIPK1, preventing NF-κB and p38MAPK activation upon TNFR1 and TLR3 stimulation [20, 21], as well as preventing apoptosis, presumably by inhibiting complex II activity. M45 is the first viral protein identified to inhibit RIPK1, but the key role of RIPK1 in viral defence suggests that there are likely to be more viruses that use this approach.
PREPARING FOR DEFEAT
Once the cellular machinery detects the viral intruder, induction of apoptosis is often the strategy of choice to inhibit viral propagation. Generally, apoptosis can be induced by three different pathways, all ultimately leading to the activation of caspases, the key executioner proteases in the apoptotic pathway. In perforin/granzyme-mediated apoptosis, caspases are directly activated by granzymes released by neighbouring immune cells that sensed viral particles by any of the above discussed mechanisms or by antigen presentation. While perforin/granzyme mediated apoptosis result in direct caspases activation, the extrinsic and the intrinsic pathways of apoptosis respectively, consist of several, highly regulated steps that will also result in activation of executioner caspases. There is an intense crosstalk between the different pathways of apoptosis and these pathways should not be considered isolated (Fig. 3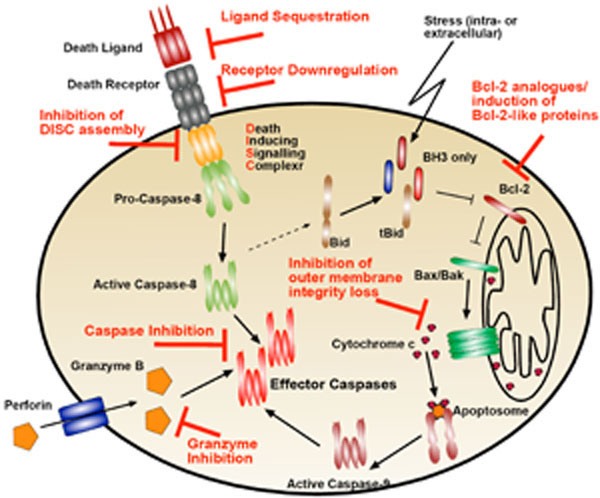 ).
).

In addition to caspase-dependent apoptosis, non-caspase-dependent forms of cell death also play important roles in the innate immune response of virus-infected cells and will also be discussed later in this article.
PERFORIN/ GRANZYME-MEDIATED APOPTOSIS
The perforin/granzyme B pathway is initiated by T-cells and natural killer (NK) cells by the release of pore-forming perforin and caspase-activating granzymes (mostly granzyme B) to a target cell. The exact mechanism of how perforin allows granzymes to enter a target cell is still under debate. The long-accepted model of pore formation is in need of some revision because the suggested pores have been shown to be too small and too transient for granzyme entry. It is likely that the insertion of perforins destabilises the target cell membrane, resulting in increased repair and endosomal uptake of granzymes [22]. Granzyme B is a serine protease that directly activates caspases, in particular the effector caspase, caspase-3, by proteolytic cleavage and thereby initiates target cell apoptosis. Viruses have been forced to inhibit perforin/granzyme-mediated cell death and the most commonly adopted tactic is direct inhibition of granzyme B. Both BHRF from Epstein Barr virus and the adenoviral L4 100 kDa protein form a stable complex with granzyme B and thereby inhibit its proteolytic activity [23, 24]. A cowpox serpin, CrmA, has also been described to inhibit granzyme B, although neither it or the closely-related ectromelia viral serpin SPI-2 inhibit its killing activity well [25].
DEATH RECEPTOR-MEDIATED APOPTOSIS
Interestingly it was the generation of mice deficient in the perforine/granzyme pathway that led to the discovery of another pathway of cell mediated apoptosis induction, the Fas-FasL pathway [26]. We now know that not only Fas (Apo-1, CD95) but also other members of the TNF superfamily such as TNFα or TRAIL can induce apoptosis in target cells upon binding to their cognate receptors, the so-called death receptors. Death ligand induced oligomerisation of the death receptors promotes the formation of the death receptor signalling complex (DISC), a platform containing death effector domains (DEDs) and the initiator caspase-8, which also contains DEDs, is recruited to this platform and activated by oligomerisation and auto-processing. Active caspase-8 can then activate effector caspases such as caspase-3.
The extrinsic pathway is a potent, rapid mechanism to drive infected cells to kill themselves. Many viruses have evolved strategies to interfere with this pathway: sequestration of death-mediating cytokines by expression of soluble receptor orthologs (pox viruses) or repression of death receptor surface expression (adenoviruses) are commonly used means of inhibiting extrinsic apoptosis [1]. Furthermore many pox viruses also encode serpins that are able to directly inhibit initiator caspase-8 (20). Gamma herpes viruses encode inhibitors of caspase-8 oligomerisation and activation called viral FLIPs (vFLIPs). vFLIPs contain two DEDs and thereby replace caspase-8 at the DISC, inhibiting the proteolytic processing of procaspase-8 to its active form. vFLIPs also interact with intracellular signal transducers via DEDs and may regulate the activation of transcription factors for anti-apoptotic proteins [27].
A different tactic is used by Epstein Barr virus [28], a B-cell-transforming virus: the protein LMP1 acts like the constitutively active TNF receptor CD40, which does not recruit caspase-8 and normally modulates TNFR1 signalling. Likewise, LMP1 engages intracellular signal transducers that result in NF-κB and Jun kinase activation and constant expression of pro-survival factors [29].
MITOCHONDRIA-MEDIATED APOPTOSIS
While the perforin/granzyme and the death receptor mediated induction of apoptosis are initiated by neighbouring (immune) cells, the Bcl-2 blockable (or mitochondria mediated/ intrinsic) pathway can be induced by almost any cell, providing it can detect the viral intruder. Pro-apoptotic proteins of the Bcl-2 family are induced or activated by cellular stress and can in turn activate the pro-apoptotic Bcl-2 family members Bax and Bak to cause a breach in the integrity of the mitochondrial outer membrane. In a healthy cell, Bax and Bak are tempered by the anti-apoptotic Bcl-2-like proteins such as Bcl-XL, Mcl-1 and Bcl-2 itself, which in turn can be antagonised by the so-called BH3-only proteins, such as Bid, Bim, Bik, Bad, Noxa or Puma (see review of G. Dewson in this issue). BH3-only proteins are often upregulated or activated by cellular stresses and therefore transduce stress signals to the apoptotic machinery. One major consequence of the loss of the mitochondrial outer membrane integrity is the release of cytochrome c from the mitochondria. Cytochrome c activates a CARD-containing platform protein called Apaf1, driving its oligomerisation, which in turn drives caspase-9 activation in a similar way to death receptors driving caspase-8 oligomerisation and activation.
Viral infection causes cellular stress by subverting the cell’s machinery to viral production and therefore viruses are very often obliged to block the mitochondrial pathway of apoptosis. A commonly used strategy is the inhibition the breach of the outer mitochondrial membrane, mediated by Bax and Bak via direct interaction with these proteins. Viral mitochondria-localised inhibitor of apoptosis (vMIA), a cytomegaloviral protein that mimics anti-apoptotic Bcl-XL, physically interacts with Bax (but not with Bak) and neutralises its pro-apoptotic function [30]. A very similar role is exhibited by various herpes virus-encoded proteins such as Herpesvirus samiri ORF16, which interacts with Bak and Bax to inhibit virus-induced apoptosis [31]. Alternatively, a variety of viral pathogens encode for functional analogues of the anti-apoptotic Bcl-2 members (also referred to as vBcl2s), such as adenoviral E1B-19kDa. E1B-19kDa prevents apoptosis induced by a variety of stimuli by preventing Bax/Bak interaction and pore formation in the mitochondrial outer membrane [32]. Another variation on the strategy to interfere with the mitochondrial pathway of apoptosis induction is the transcriptional upregulation of anti-apoptotic Bcl-2 family members. Such an example is the human T-cell lymphoma virus (HTLV) encoded Tax protein, which activates the Bcl-XL promoter while repressing Bax expression [33].
CASPASE-INDEPENDENT CELL DEATH
A form of caspase-independent cell death is autophagy, a mechanism that was initially associated with the starvation of single cells upon nutrient starvation. A more adequate view is, however, to consider autophagy as an important remodelling process, which, in certain cases, can result in cell death. During autophagy, a cell degrades long-lived proteins and entire organelles in autophagosomes, double-membrane vesicles within the cytoplasm. Cell death by autophagy is readily used as an anti-viral mechanism, most prominently in plants. As expected, the anti-viral properties of autophagy are also subverted by some viruses; the autophagosome is used as a niche for replication (coronavirus, mouse hepatitis virus, rhinovirus), and it serves as an excellent release mechanism of some non-enveloped RNA viruses (polioviruses). A role for autophagy in viral defence has been further highlighted recently by a study showing the autophagy proteins Atg5 and Atg12 regulate the Rig-I mediated dsRNA detection pathway [34].
Another recently defined form of caspase-independent cell death is programmed necrosis or necroptosis. Necroptosis can be considered as an evasion mechanism that is activated when the default program (apoptosis) is blocked, for instance, by viral or chemical blockage of caspases. A central role in the regulation of necroptosis is associated with the proteins RIPK1 and RIPK3, which are important factors in the initiation of an anti-viral response by dimerising and activating each other, leading to necrosis [35]. RIPK1 also plays an important role in this complex, since inhibition of RIPK1 kinase activity rescues certain cells from TNFα-induced cell death (reviewed in ref [2]).
The previously mentioned CMV protein M45 antagonises RIPK1 and RIPK3 interactions during viral infection and blocks necrosis. Consequently TNFα fails to induce NF-κB and TNFα-induced cell death is blocked [36]. The reason for this phenomenon is biologically fascinating and a prime example of the arms race between virus and host cell: CMV harbours a caspase-8 inhibiting protein (M36) and therefore blocks classic apoptosis. The default program of cell death would therefore be necroptosis; however, due to M45-mediated RIPK1 and RIPK3 inhibition, this form of cell death is also blocked and CMV is able to keep the host cell alive to replicate its genome [37].
THE LAST LINE OF DEFENCE
Desperate as it may seem, there are a few virus-encoded proteins that appear to target executioner caspases and therefore prolong the host cell’s lifespan enough to ensure its own replication. One such protein is p35 encoded by baculoviruses, which acts as a pseudosubstrate and traps caspases in a detergent-resistant complex, due to a conformational change upon cleavage. Therefore, even activated effector caspases are blocked and the ongoing cellular destruction program is permanently inhibited or slowed down [27].
A more efficient strategy than inhibition of effector caspases is to target initiator caspases, either on their activation or when already activated. CrmA from the cowpox virus blocks initiator caspase-8, thereby inhibiting the extrinsic apoptotic pathway and blocks inflammatory caspase-1, thereby limiting the production of interleukin-1ß and dampening the immune response [27]. Inhibitor of apoptosis proteins (IAPs) were initially discovered in baculoviruses and act in a similar way as CrmA or p35. The discovery of viral IAPs led to the identification of their cellular homologues, which are now considered to be promising targets for cancer therapy in humans. The prototype of cellular IAPs is DIAP1, which efficiently inhibits cell death in the fruit fly by binding to and targeting the caspase-9 orthologue DRONC to proteasomal degradation by polyubiquitinylation [38] [39]. Thereby the ‘Sword of Damocles’, which in this case is the aberrantly present, but yet inactive, caspases is removed. In contrast, the mammalian X-linked IAP (XIAP) directly inhibits caspases at physiological concentrations [40]. A similar mechanism of action was proposed for the mammalian cellular IAPs cIAP-1 and cIAP-2, but it was recently shown that these IAPs are not able to directly inhibit caspases [41].
A fascinating strategy is used by vaccinia viruses, which use mimicry of an apoptotic cell to invade their hosts [42]. Upon infection, the virus induces cellular blebbing with many features of blebbing observed after apoptosis induction such as exposure of phosphatidylserine. These potentially apoptotic bodies are then pinocytosed and the virus got a free ride into the cell it intends to infect, without induction of an immune response.
It is worth to mention, that complete inhibition of host cell death by the virus is not always desired since release of virus particles can be dependent on the disintegration of the respective host cells. Therefore many viral strategies interfering with the host cell death machinery result in a delay rather than a complete inhibition of host cell death. To ensure proper function of this release mechanism, viruses also developed a wide variety of strategies to ensure death of the host cell, a mechanism which is not discussed in this article but certainly contributes as much to the success of viruses as the discussed mechanisms to inhibit or delay cell death.
WHO STRUCK FIRST?
The fact that viral pathogens with their very limited genome size have developed a whole arsenal of anti-apoptotic proteins highlights that evasion from host cell apoptosis is required for viral survival. Apoptosis has been shown to be important during embryonic development, tissue homeostasis and in the regulation of the immune system, and aberrant apoptosis regulation is associated with a wide variety of diseases such as cancer and autoimmune diseases (nicely reviewed by M. Ditzel in this issue). But the question remains whether the driving force to develop and refine apoptosis as a self-destruction program has been due to pathogenic pressure. Whatever the answer, the arms race between virus and host in this suicidal struggle continues to amaze with its complexity and sophistication and shines new light on this morbidly fascinating process.