- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
A New Structure-Based QSAR Method Affords both Descriptive and Predictive Models for Phosphodiesterase-4 Inhibitors
Xialan Dong , Weifan Zheng*
Abstract
We describe the application of a new QSAR (quantitative structure-activity relationship) formalism to the analysis and modeling of PDE-4 inhibitors. This new method takes advantage of the X-ray structural information of the PDE-4 enzyme to characterize the small molecule inhibitors. It calculates molecular descriptors based on the matching of their pharmacophore feature pairs with those (the reference) of the target binding pocket. Since the reference is derived from the X-ray crystal structures of the target under study, these descriptors are target-specific and easy to interpret. We have analyzed 35 indole derivative-based PDE-4 inhibitors where Partial Least Square (PLS) analysis has been employed to obtain the predictive models. Compared to traditional QSAR methods such as CoMFA and CoMSIA, our models are more robust and predictive measured by statistics for both the training and test sets of molecules. Our method can also identify critical pharmacophore features that are responsible for the inhibitory potency of the small molecules. Thus, this structure-based QSAR method affords both descriptive and predictive models for phosphodiesterase-4 inhibitors. The success of this study has also laid a solid foundation for systematic QSAR modeling of the PDE family of enzymes, which will ultimately contribute to chemical genomics research and drug discovery targeting the PDE enzymes.
Article Information
Identifiers and Pagination:
Year: 2008Volume: 2
First Page: 29
Last Page: 39
Publisher Id: CCGTM-2-29
DOI: 10.2174/1875397300802010029
Article History:
Received Date: 30/7/2008Revision Received Date: 2/9/2008
Acceptance Date: 2/9/2008
Electronic publication date: 6/11/2008
Collection year: 2008
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Department of Pharmaceutical Sciences, BRITE Institute, North Carolina Central, University, 1801 Fayetteville Street, Durham, NC 27707, USA; Tel: 919 530 6752; Fax: 919 530 6600; E-mail: wzheng@nccu.edu
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 30-7-2008 |
Original Manuscript | A New Structure-Based QSAR Method Affords both Descriptive and Predictive Models for Phosphodiesterase-4 Inhibitors | |
INTRODUCTION
The cyclic nucleotide phosphodiesterases (or PDE’s) comprise a group of enzymes that degrade the phosphodiester bond in the second messenger molecules cAMP and cGMP. They regulate the localization, duration, and amplitude of cyclic nucleotide signaling within subcellular domains. Thus, PDE’s are important regulators of signal transduction mediated by these second messenger molecules [1Torphy TJ. Phosphodiesterase isozymes: molecular targets for novel antiasthma agents Am J Respir Crit Care Med 1998; 157(2): 351-70.-3Beavo JA, Conti M, Heaslip RJ. Multiple cyclic nucleotide phosphodiesterases Mol Pharmacol 1994; 46(3): 399-405.].
According to their sequence homology, substrate specificity and pharmacological properties, the PDE enzymes are classified into 11 families (i.e., PDE1 - PDE11) [2Soderling SH, Beavo JA. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions Curr Opin Cell Biol 2000; 12(2): 174-9.]. Even though different subtypes of this super-family are functionally related, they do have different substrate specificities. For example, PDE-4, PDE-7 and PDE-8 are cAMP selective, whereas PDE-5, PDE-6 and PDE-9 are cGMP selective. Other members of this family (e.g., PDE-1, PDE-2, PDE-3, PDE-10 and PDE-11) can hydrolyse both cAMP and cGMP.
Phosphodiesterases have proven to be a therapeutically important class of drug targets. Sildenafil is an inhibitor of cGMP-specific phosphodiesterase (PDE-5) and used to treat erectile dysfunction (ED) [4McCullough AR, Steidle CP, Klee B, Tseng LJ. Randomized, double-blind, crossover trial of sildenafil in men with mild to moderate erectile dysfunction: efficacy at 8 and 12 hours postdose Urology 2008; 71(4): 686-92.]. Inhibitors of PDE-4 have been explored as potential drugs for chronic obstructive pulmonary disorder (COPD) [5Brown WM. Treating COPD with PDE 4 inhibitors Int J Chron Obstruct Pulmon Dis 2007; 2(4): 517-33.]. Inhibitors of other PDE enzymes are being explored for coronary heart disease [6Guazzi M, Samaja M. The role of PDE5-inhibitors in cardiopulmonary disorders: from basic evidence to clinical development Curr Med Chem 2007; 14(20): 2181-91.], dementia, depression, and schizophrenia [7Halene TB, Siegel SJ. PDE inhibitors in psychiatry--future options for dementia, depression and schizophrenia? Drug Discov Today 2007; 12(19-20): 870-.]. Thus, there are continuing interests in discovering novel inhibitors of PDE enzymes that are potent and selective for specific subtypes.
We have been interested in modeling the inhibitors of PDE-4 due to their potential in treating neurodegenerative diseases and cognitive disorders. PDE-4 selectively hydrolyzes cAMP and has been mostly targeted for the treatment of inflammation, and in particular chronic obstructive pulmonary disorder (COPD) [5Brown WM. Treating COPD with PDE 4 inhibitors Int J Chron Obstruct Pulmon Dis 2007; 2(4): 517-33.]. However, selective phosphodiesterase-4 (PDE-4) inhibitors have been demonstrated recently as novel agents to treat memory deficit [8Ghavami A, Hirst WD, Novak TJ. Selective phosphodiesterase (PDE)-4 inhibitors: a novel approach to treating memory deficit? Drugs R D 2006; 7(2): 63-71.] and neurodegeneration [9Chen RW, Williams AJ, Liao Z, Yao C, Tortella FC, Dave JR. Broad spectrum neuroprotection profile of phosphodiesterase inhibitors as related to modulation of cell-cycle elements and caspase-3 activation Neurosci Lett 2007; 418(2): 165-9.]. Preclinical studies indicate that these PDE-4 inhibitors can counteract deficits in long-term memory caused by pharmacological agents, aging or over-expression of mutant forms of human amyloid precursor proteins [9Chen RW, Williams AJ, Liao Z, Yao C, Tortella FC, Dave JR. Broad spectrum neuroprotection profile of phosphodiesterase inhibitors as related to modulation of cell-cycle elements and caspase-3 activation Neurosci Lett 2007; 418(2): 165-9.]. Although a number of major advances have been made with respect to PDE-4 inhibition, most of the PDE-4 inhibitors have unacceptable side effects, particularly nausea and emesis [10Banner KH, Trevethick MA. PDE4 inhibition: a novel approach for the treatment of inflammatory bowel disease Trends Pharmacol Sci 2004; 25(8): 430-6., 11Burnouf C, Pruniaux MP. Recent advances in PDE4 inhibitors as immunoregulators and anti-inflammatory drugs Curr Pharm Des 2002; 8(14): 1255-96.]. Thus, it is critically important to understand the structural determinants of potent and selective PDE-4 inhibitors, so that we can rationally design new molecules that minimize the undesirable side effects.
Most PDE-4 inhibitors can be classified into three structural families: rolipram-related compounds, xanthine derivatives and nitraquazone analogues [12Ducrot P, Andrianjara CR, Wrigglesworth R. CoMFA and CoMSIA 3D-quantitative structure-activity relationship model on benzodiazepine derivatives, inhibitors of phosphodiesterase IV J Comput-Aided Mol Des 2001; 15(9): 767-85.]. This structural diversity suggests that the binding site of the PDE-4 enzyme presents a number of alternative pharmacophores that are capable of interacting with different classes of small molecule inhibitors. Many investigators have tried to understand the structural basis for PDE-4 inhibition (both potency and selectivity) by constructing 3D QSAR (quantitative structure-activity relationship) models based on known PDE-4 inhibitors [12Ducrot P, Andrianjara CR, Wrigglesworth R. CoMFA and CoMSIA 3D-quantitative structure-activity relationship model on benzodiazepine derivatives, inhibitors of phosphodiesterase IV J Comput-Aided Mol Des 2001; 15(9): 767-85.-16Yang Z, Sun P. 3D-QSAR study of potent inhibitors of phosphodiesterase-4 using a CoMFA approach Int J Mol Sci 2007; 8: 714-22.]. Even though these models provided preliminary pharmacophoric insights into PDE-4 inhibitors, the absence of the 3D structure of the target enzyme (i.e., PDE-4) in these models poses a major limitation on the reliability and interpretability of these models. Other traditional 2D QSAR analyses often result in only statistical models with little to offer in terms of structural insights that the medicinal chemist can use to optimize the inhibitors [12Ducrot P, Andrianjara CR, Wrigglesworth R. CoMFA and CoMSIA 3D-quantitative structure-activity relationship model on benzodiazepine derivatives, inhibitors of phosphodiesterase IV J Comput-Aided Mol Des 2001; 15(9): 767-85., 17Brogle KC, Gund T, Kyle DJ. Improving synthetic efficiency using the computational prediction of biological activity Comb Chem High Throughput Screen 2006; 9(2): 103-3.]. Thus, it is highly desirable to develop structure-based, both descriptive and predictive models.
Here, we describe the application of a new QSAR method to the analysis and modeling of PDE-4 inhibitors. This method takes advantage of the X-ray structural information of the target enzyme (i.e., PDE4) to characterize its small molecule inhibitors. Other groups have published different methods to conduct conceptually similar QSAR analysis on other targets, notably the work by Hopfinger’s group and Cruciani’s group [18Santos-Filho OA, Hopfinger AJ. Structure-based QSAR analysis of a set of 4-hydroxy-5,6-dihydropyrones as inhibitors of HIV-1 protease: an application of the receptor-dependent (RD) 4D-QSAR formalism J Chem Inf Model 2006; 46(1): 345-54.-22Baroni M, Cruciani G, Sciabola S, Perruccio F, Mason JS. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): theory and application J Chem Inf Model 2007; 47(2): 279-94.]. Our method derives the pharmacophoric centers from the 3D structure of PDE-4’s binding pocket, and uses them as pharmacophore reference. It then generates structure-based pharmacophore key (SB-PPK) descriptors for the inhibitors based on their pharmacophoric matching patterns with the above reference. We have analyzed 35 indole derivative-based PDE-4 inhibitors [15Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38(11-12): 975-82.]. Partial Least Square (PLS) analysis has been employed to obtain the predictive models measured by both the training set and test set statistics [23Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models J Comput-Aided Mol Des 2003; 17(2-4): 241-53.]. We will compare our results with previous studies of the same data set by more conventional methods and highlight some novel aspects of our approach.
METHODS AND MATERIALS
Dataset
35 indole derivative-based PDE-4 inhibitors [15Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38(11-12): 975-82.] were used in this study. The molecular structures and their biological activities against PDE-4 are given in Table 1. The compounds contain an indole moiety which replaces the “rolipram-like” 3-methoxy-4-cyclopentoxy motif. The activities were determined from measurement of serum TNF-α levels in LPS challenged mice (mouse endotoxemia model) [24Hulme C, Moriarty K, Miller B, et al. The synthesis and biological evaluation of a novel series of indole PDE4 inhibitors I Bioorg Med Chem Lett 1998; 8(14): 1867-72.].
This dataset covers a variety of molecules with various levels of inhibitory activities against PDE-4. Fig. (1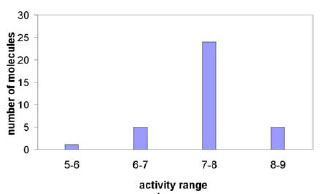 ) shows the activity distribution of the 35 indole derivatives. To develop and rigorously validate a QSAR model, 28 molecules were selected as the training set, and the remaining 7 molecules were held out as the test set. The specific selection of the training and test sets was determined by either randomly splitting or clustering-based splitting of the original dataset.
) shows the activity distribution of the 35 indole derivatives. To develop and rigorously validate a QSAR model, 28 molecules were selected as the training set, and the remaining 7 molecules were held out as the test set. The specific selection of the training and test sets was determined by either randomly splitting or clustering-based splitting of the original dataset.
The 3D conformations of all the inhibitors were generated using the OMEGA 2.0 program available from OpenEye Scientific (OMEGA 2.0, OE Scientific, NM, USA). In this study, only the best conformer of each molecule was used to generate the pharmacophore points.
The Overall Work Flow of the QSAR Method
Fig. (2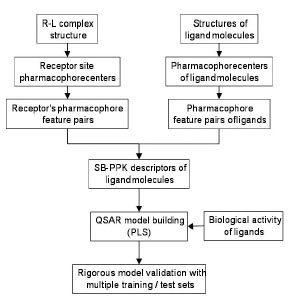 ) depicts the overall work flow of the structure-based QSAR method employed in this study. The novel aspect of this method is the way by which it generates the molecular descriptors by the SB-PPK (Structure-based Pharmacophore Key) program. Unlike conventional 2D or 3D molecular descriptor generation methods where ONLY the structural information of the inhibitor molecules is used to characterize the molecules, the SB-PPK program uses the structural information of the target binding site to measure the inhibitor molecules. Thus, target-specific molecular descriptors are generated, which reflects not only the inhibitor molecules but also the way these molecules interact with the binding site of the target. The general process of SB-PPK involves: 1) extraction of pharmacophore features from the target binding site; 2) perception of pharmacophore features of small molecules; and 3) generation of the structure-based pharmacophore key (SB-PPK) descriptors. Specifically, it derives the pharmacophoric centers from the 3D structure of a protein-ligand (R-L) complex, and uses them to generate the pharmacophore reference. It then generates structure-based pharmacophore key (SB-PPK) descriptors for the inhibitor molecules based on their pharmacophoric matching patterns with the above reference. Once molecular descriptors are generated, PLS (Partial Least Square) method is employed to establish the relationship between these molecular descriptors and the activity of the inhibitors. Details of the individual steps are described as follows.
) depicts the overall work flow of the structure-based QSAR method employed in this study. The novel aspect of this method is the way by which it generates the molecular descriptors by the SB-PPK (Structure-based Pharmacophore Key) program. Unlike conventional 2D or 3D molecular descriptor generation methods where ONLY the structural information of the inhibitor molecules is used to characterize the molecules, the SB-PPK program uses the structural information of the target binding site to measure the inhibitor molecules. Thus, target-specific molecular descriptors are generated, which reflects not only the inhibitor molecules but also the way these molecules interact with the binding site of the target. The general process of SB-PPK involves: 1) extraction of pharmacophore features from the target binding site; 2) perception of pharmacophore features of small molecules; and 3) generation of the structure-based pharmacophore key (SB-PPK) descriptors. Specifically, it derives the pharmacophoric centers from the 3D structure of a protein-ligand (R-L) complex, and uses them to generate the pharmacophore reference. It then generates structure-based pharmacophore key (SB-PPK) descriptors for the inhibitor molecules based on their pharmacophoric matching patterns with the above reference. Once molecular descriptors are generated, PLS (Partial Least Square) method is employed to establish the relationship between these molecular descriptors and the activity of the inhibitors. Details of the individual steps are described as follows.
 |
Fig. (2) The overall flowchart of the SB-PPK QSAR Method. |
Deriving Pharmacophore Feature Pairs from Ligand-Receptor (R-L) Complex
The LigandScout program [25Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters J Chem Inf Model 2005; 45(1): 160-9.] was first employed to derive structure-based pharmacophore centers (Fig. 3a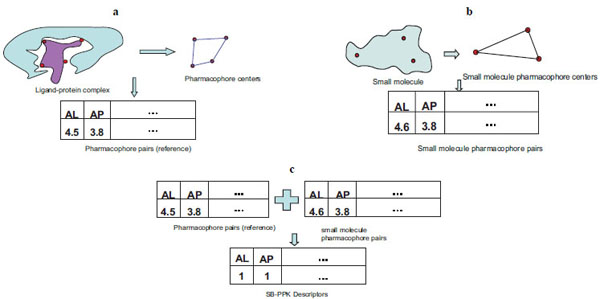 ). The advanced 3D pharmacophore model option of the LigandScout program was used, which allows multiple pharmacophore feature types on the same heavy atom. The pharmacophore feature types used in this study include hydrogen bond donor (D), hydrogen bond acceptor (A), positively charged (P), negatively charged (N), and lipophilic (L) centers. Pharmacophore feature pairs are then generated by making all possible pair-wise combinations of the above pharmacophore centers. A specific pharmacophore feature pair is determined by both the feature types involved and the distance between the two centers. For example, AL in Fig. (3a) indicates a hydrogen bond acceptor (A) and lipophilic (L) feature pair; and the distance between them is 4.5 A. Another example, AP, means a hydrogen bond acceptor (A) and a positive (P) feature pair and their inter-feature distance is 3.8 A. All possible feature pairs are generated, and this recorded information reflects the pharmacophoric characteristics of the target binding site, and will be used as the reference for generating the SB-PPK descriptors of the inhibitor molecules. To avoid confusion with traditional pharmacophore fingerprints, we note that we do not need “distance binning” to capture different possible distance ranges because the pharmacophore features pairs are derived from the 3D structure of the target protein. Thus, all the feature pairs have defined distances.
). The advanced 3D pharmacophore model option of the LigandScout program was used, which allows multiple pharmacophore feature types on the same heavy atom. The pharmacophore feature types used in this study include hydrogen bond donor (D), hydrogen bond acceptor (A), positively charged (P), negatively charged (N), and lipophilic (L) centers. Pharmacophore feature pairs are then generated by making all possible pair-wise combinations of the above pharmacophore centers. A specific pharmacophore feature pair is determined by both the feature types involved and the distance between the two centers. For example, AL in Fig. (3a) indicates a hydrogen bond acceptor (A) and lipophilic (L) feature pair; and the distance between them is 4.5 A. Another example, AP, means a hydrogen bond acceptor (A) and a positive (P) feature pair and their inter-feature distance is 3.8 A. All possible feature pairs are generated, and this recorded information reflects the pharmacophoric characteristics of the target binding site, and will be used as the reference for generating the SB-PPK descriptors of the inhibitor molecules. To avoid confusion with traditional pharmacophore fingerprints, we note that we do not need “distance binning” to capture different possible distance ranges because the pharmacophore features pairs are derived from the 3D structure of the target protein. Thus, all the feature pairs have defined distances.

Perception of Pharmacophore Feature Pairs for a Ligand
The LigandScout program[25Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters J Chem Inf Model 2005; 45(1): 160-9.] is used to perceive the pharmacophoric groups on small organic molecules (Fig. 3b). All pair-wise combinations of the perceived pharmacophore centers are checked for the feature types and interfeature distances. In the example given in Fig. (3b), there is an AL pair and an AP pair, and the inter-feature distances for each pair are 4.6 A and 3.8 A, respectively.
Generation of Structure-Based Pharmacophore Key (SB-PPK) Descriptors
Once the pharmacophore feature pairs for both the receptor and ligand molecules are generated, we can compare them to determine pattern matches. For example, Fig. (3c) shows that AL’s and AP’s from the receptor’s pharmacophore feature pairs (the reference) and small molecule’s pharmacophore pairs match with each other. Thus, the corresponding positions for AL and AP are increased by 1 count. Both fingerprint-like binary descriptors and feature pair counts descriptors can be generated for a given ligand.
Statistical Analysis with Partial Least Square (PLS) Method
The relationship between the molecular descriptors and the biological activities is modeled by the PLS (Partial Least Square) method [26Hokuldsson A. A PLS regression methods J Chemometr 1988; 2: 211-8.]. Specifically, the PLS implementation in the MOE package (Chemical Computing Group, Montreal, Canada) was used in this study. Other types of machine learning methods can be used as well to analyze the dataset.
Validation of the QSAR Models
Model validation is a critical step in any QSAR study. As shown in Fig. (4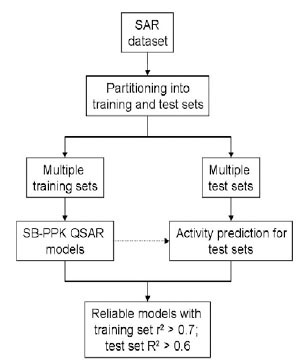 ), we have adopted a standard workflow, similar to what Tropsha group advocate [23Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models J Comput-Aided Mol Des 2003; 17(2-4): 241-53.], to build robustly predictive QSAR models. Rational splitting of the dataset into training and test sets as well as obtaining the statistics for both the training set and test sets is critical in this model validation protocol. Thus, a given dataset is split into multiple pairs of training and test sets. Both random shuffling and clustering-based [27Labute P. Quasar-Cluster: A different view of molecular clustering Chem Comp Group J 2002 [Jan 5, 2008]; http://www.chemcomp.com/journal/cluster.htm] splitting of the data set can be used for dataset partitioning. In the random shuffling approach, the first 7 molecules were held out as the test set after the order of molecules in the data set was randomly shuffled. In the clustering-based partitioning approach, the dataset was split in such a way that both the training set (28 molecules) and the test set (7 molecules) had the most coverage of the clusters. For each split, PLS was used to analyze the training set in order to build a PLS regression model. Regression coefficients were then calculated for both the training set (r2) and the test set (R2). While the former reflects the model quality as measured on the training set, the latter (R2) is a more robust indicator of the predictive power of the model. In the end, only those model(s) with both coefficients greater than set thresholds are retained as the final model(s).
), we have adopted a standard workflow, similar to what Tropsha group advocate [23Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models J Comput-Aided Mol Des 2003; 17(2-4): 241-53.], to build robustly predictive QSAR models. Rational splitting of the dataset into training and test sets as well as obtaining the statistics for both the training set and test sets is critical in this model validation protocol. Thus, a given dataset is split into multiple pairs of training and test sets. Both random shuffling and clustering-based [27Labute P. Quasar-Cluster: A different view of molecular clustering Chem Comp Group J 2002 [Jan 5, 2008]; http://www.chemcomp.com/journal/cluster.htm] splitting of the data set can be used for dataset partitioning. In the random shuffling approach, the first 7 molecules were held out as the test set after the order of molecules in the data set was randomly shuffled. In the clustering-based partitioning approach, the dataset was split in such a way that both the training set (28 molecules) and the test set (7 molecules) had the most coverage of the clusters. For each split, PLS was used to analyze the training set in order to build a PLS regression model. Regression coefficients were then calculated for both the training set (r2) and the test set (R2). While the former reflects the model quality as measured on the training set, the latter (R2) is a more robust indicator of the predictive power of the model. In the end, only those model(s) with both coefficients greater than set thresholds are retained as the final model(s).
 |
Fig. (4) Workflow for rigorous validation of QSAR models. |
RESULTS AND DISCUSSION
Pharmacophore Features of the PDE-4 Binding Site
As described before (Cf. Method), LigandScout was used to analyze the interactions between PDE-4 inhibitors and the binding site of PDE-4, based on the X-ray structure of the inhibitor-bound PDE-4 (1xon.pdb). This analysis has revealed nine pharmacophore centers. The 3D rendering and the 2D depiction of the interactions are shown in Fig. (5a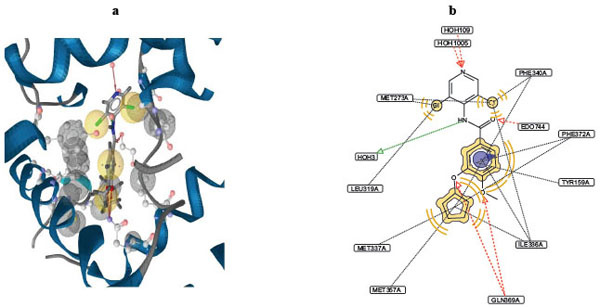 ) and Fig. (5b), respectively. The following occupies the four hydrophobic centers: the cyclopentane ring, two chlorine atoms on the pyridine ring, and the central benzene ring. The cyclopentane ring interacts with hydrophobic residues in the PDE-4 binding pocket: MET337, MET357, PHE340, PHE372, and ILE336. Chlorines on the pyridine ring interact with hydrophobic residues MET273, PHE340, ILE336, and LEU319, and the central benzene ring interacts with ILE336, PHE372 and TYR159. The amide nitrogen occupies a hydrogen bond donor center, which interacts with a water molecule that serves as a bridge to the enzyme’s binding pocket. Four hydrogen bond acceptor centers are identified and they are occupied by the following atoms/groups: the methoxy oxygen on the benzene ring, amide oxygen, pyridine nitrogen, and the ether oxygen (connecting the benzene and the cyclopentane rings). The methoxy oxygen and the ether oxygen atoms both interact with GLN369. The amide oxygen and the pyridine nitrogen interact with the small molecule EDO744 and a water molecule, respectively.
) and Fig. (5b), respectively. The following occupies the four hydrophobic centers: the cyclopentane ring, two chlorine atoms on the pyridine ring, and the central benzene ring. The cyclopentane ring interacts with hydrophobic residues in the PDE-4 binding pocket: MET337, MET357, PHE340, PHE372, and ILE336. Chlorines on the pyridine ring interact with hydrophobic residues MET273, PHE340, ILE336, and LEU319, and the central benzene ring interacts with ILE336, PHE372 and TYR159. The amide nitrogen occupies a hydrogen bond donor center, which interacts with a water molecule that serves as a bridge to the enzyme’s binding pocket. Four hydrogen bond acceptor centers are identified and they are occupied by the following atoms/groups: the methoxy oxygen on the benzene ring, amide oxygen, pyridine nitrogen, and the ether oxygen (connecting the benzene and the cyclopentane rings). The methoxy oxygen and the ether oxygen atoms both interact with GLN369. The amide oxygen and the pyridine nitrogen interact with the small molecule EDO744 and a water molecule, respectively.

Pharmacophore Reference Generated from the PDE-4 Binding Site
Based on the above-generated pharmacophore features, we created the pharmacophore reference in terms of pharmacophore feature pairs (Cf. Method). Each structure based descriptor is determined by the feature types involved in a pharmacophore feature pair and the distance between the two feature centers. Table 2 shows the pharmacophore feature pairs (the reference) derived from the PDE4-inhibitor complex (1xon.pdb). It has six AA pairs with inter-feature distances ranging from 6.34 to 10.69 A. Four AD pairs with inter-feature distances ranging from 2.29 to 6.50 A. Likewise, there are sixteen AL pairs, four DL pairs and six LL pairs with different inter-feature distances. Together, this set of feature pairs uniquely characterizes the binding pocket of the PDE-4 enzyme, and serves as a reference with which candidate inhibitor molecules can be compared to generate their molecular descriptors.
Structure-based Pharmacophore Key (SB-PPK) Descriptors of PDE-4 Inhibitors
The descriptors of the PDE-4 inhibitors were determined based on the matches between the feature pairs in the reference and those in each individual molecule. Fig. (6a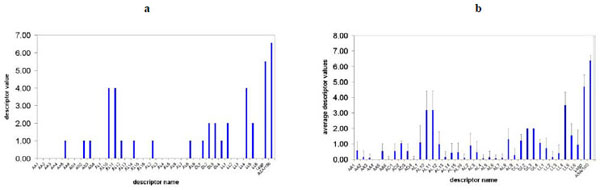 ) shows the values of calculated SB-PPK descriptors of PDE-4 inhibitor 3 (mol.3 in Table 1). The average descriptor values over the dataset of 35 PDE-4 inhibitors are presented in Fig. (6b). In inhibitor 3, the value of AL10 is 4, which indicates 4 possible AL10 (inter-feature distance 3.98) matches between the feature pairs of inhibitor 3 and those of the binding site reference. Obviously, different molecules have different number of matches between different feature pairs. In Fig. (6b), the error bars show the variations of each descriptor value around the averages, amongst the different inhibitors in the dataset under study. Those descriptors with larger variations tend to have more important roles in QSAR models.
) shows the values of calculated SB-PPK descriptors of PDE-4 inhibitor 3 (mol.3 in Table 1). The average descriptor values over the dataset of 35 PDE-4 inhibitors are presented in Fig. (6b). In inhibitor 3, the value of AL10 is 4, which indicates 4 possible AL10 (inter-feature distance 3.98) matches between the feature pairs of inhibitor 3 and those of the binding site reference. Obviously, different molecules have different number of matches between different feature pairs. In Fig. (6b), the error bars show the variations of each descriptor value around the averages, amongst the different inhibitors in the dataset under study. Those descriptors with larger variations tend to have more important roles in QSAR models.
 |
Fig. (6) a. The SB-PPK descriptor values of a PDE4 inhibitor (mol.3 of Table 1). b. The SB-PPK descriptor values averaged over the 35 PDE4 Inhibitors. |
QSAR Model Building with Partial Least Square (PLS) Method
We follow the workflows outlined in Fig. (2) and Fig. (4) to conduct the QSAR model building and model validation. A variety of factors that may have significant effect on QSAR model quality have been thoroughly examined and discussed below. The quality of the final model is compared with previous work.
Effect of Number of Principle Components on the Quality of QSAR Models
To obtain predictive QSAR models, we systematically performed PLS analysis with different number of principle components used in the regression model. Fig. (7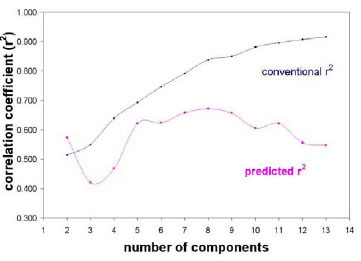 ) shows how the number of principle components affects the quality of the resulting models for the best partitioning of training and test sets. The conventional correlation coefficient r2 obtained for the training set monotonically increases while the predictive R2 obtained for the testing set reaches a plateau before it starts to decrease. When the number of principle components (PC) is less than 3, the resulting QSAR models are not predictive for either the training set or the test set. When the number of principle components is over 8, the predictive ability for the test set starts to go down. This indicates potential over-training at higher number of principle components. According to our model validation protocol (Fig. 4), the models with the number of principle components between 5 and 8 are predictive for both the training and the test sets. The best models should be a trade-off between conventional r2 and the predictive R2. Also, to compare with other literature work which uses number of principle components of 6 [15Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38(11-12): 975-82.], we decided to use 6 components to build the final best models.
) shows how the number of principle components affects the quality of the resulting models for the best partitioning of training and test sets. The conventional correlation coefficient r2 obtained for the training set monotonically increases while the predictive R2 obtained for the testing set reaches a plateau before it starts to decrease. When the number of principle components (PC) is less than 3, the resulting QSAR models are not predictive for either the training set or the test set. When the number of principle components is over 8, the predictive ability for the test set starts to go down. This indicates potential over-training at higher number of principle components. According to our model validation protocol (Fig. 4), the models with the number of principle components between 5 and 8 are predictive for both the training and the test sets. The best models should be a trade-off between conventional r2 and the predictive R2. Also, to compare with other literature work which uses number of principle components of 6 [15Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38(11-12): 975-82.], we decided to use 6 components to build the final best models.
 |
Fig. (7) Quality of the QSAR models depends on the number of principle components (PC) employed. |
Effect of Dataset Partitioning on QSAR Model Quality
To build high quality, predictive QSAR models, we systematically performed dataset splitting into training and test sets using both random shuffling and clustering-based splitting methods. For each partitioning approach, we obtained ten different training/test sets, and built 90 different QSAR models with the number of principal components ranging from 2 to 10. Of all 180 QSAR models, the conventional correlation coefficient r2 ranges from 0.69 to 0.89, and the predictive R2 values range from 0.05 to 0.75. The splitting which renders the highest r2 and R2 for corresponding models with number of principle components between 5 and 8 is selected as the partitioning of training and test sets to build the final QSAR models.
The Best SB-PPK Models have Better Predictive Ability than those Obtained with other Methods
Fig. (8a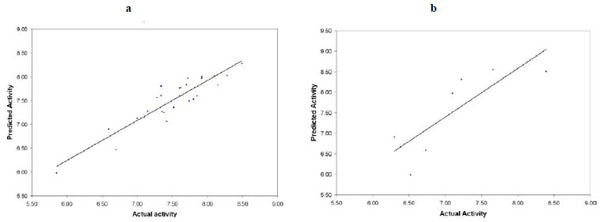 ) and Fig. (8b) show the predictive quality of the best QSAR models for PDE-4 inhibitors built with the SB-PPK descriptors and PLS method. Table 3 present the comparison between our best models with those of other methods. We have compared the results obtained with CoMFA [15Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38(11-12): 975-82.], CoMSIA [15Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38(11-12): 975-82.], 2D QSAR (using MOE package) and SB-PPK (our own method). All methods use PLS as the modeling tool with number of principle components set at 6. Both CoMFA and CoMSIA afforded highly predictive models based on the training set; however, the predictive R2 for the test set are both significantly lower than that for the training set. Our method, on the other hand, afforded much more balanced predictive quality, for both the training set and the test set. Thus, based on the reported correlation coefficients (for both training and test sets), we believe our method (SB-PPK plus the specific workflow process) afforded the best model both in terms of predictive ability for the training set and, more importantly, in terms of the predictive power on the test set.
) and Fig. (8b) show the predictive quality of the best QSAR models for PDE-4 inhibitors built with the SB-PPK descriptors and PLS method. Table 3 present the comparison between our best models with those of other methods. We have compared the results obtained with CoMFA [15Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38(11-12): 975-82.], CoMSIA [15Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38(11-12): 975-82.], 2D QSAR (using MOE package) and SB-PPK (our own method). All methods use PLS as the modeling tool with number of principle components set at 6. Both CoMFA and CoMSIA afforded highly predictive models based on the training set; however, the predictive R2 for the test set are both significantly lower than that for the training set. Our method, on the other hand, afforded much more balanced predictive quality, for both the training set and the test set. Thus, based on the reported correlation coefficients (for both training and test sets), we believe our method (SB-PPK plus the specific workflow process) afforded the best model both in terms of predictive ability for the training set and, more importantly, in terms of the predictive power on the test set.

Critical Pharmacophore Features have been Revealed by the SB-PPK QSAR Analysis
One of the important tasks of QSAR analysis is to identify critical features that are responsible for explaining the SAR trend in the dataset. In order to do so, we define a score for each SB-PPK descriptor based on the magnitude of the coefficient for that descriptor in the PLS regression models. Specifically, we define the score according to equations 1 and 2.
Score (i) is the score of a given descriptor contributed by a particular model, i, and rank is the rank order of a particular descriptor according to the magnitude of its coefficient in that PLS model. The summation is conducted over all the predictive models obtained. In this study, we base the score on four predictive PLS models, corresponding to the number of principal components 5, 6, 7, and 8. Fig. (9a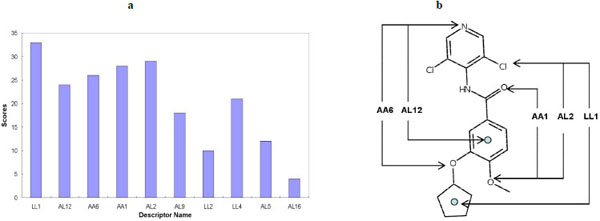 ) shows that descriptor LL1, AL12, AA6, AA1 and AL2 are relatively important in the QSAR models. These descriptors correspond to the pharmacophore feature pairs depicted in Fig. (9b). Thus, the SB-PPK descriptor based QSAR analysis can reveal critical features that correspond to the pharmacophore features of the target binding pocket. This descriptive ability is very useful and often desired by the medicinal chemist working on compound optimizations.
) shows that descriptor LL1, AL12, AA6, AA1 and AL2 are relatively important in the QSAR models. These descriptors correspond to the pharmacophore feature pairs depicted in Fig. (9b). Thus, the SB-PPK descriptor based QSAR analysis can reveal critical features that correspond to the pharmacophore features of the target binding pocket. This descriptive ability is very useful and often desired by the medicinal chemist working on compound optimizations.
 |
Fig. (9) a. Descriptor significance scores for the most important SB-PPK descriptors. b. Mapping the significant pharmacophore feature pairs according to the descriptor significance scores. |
CONCLUSIONS
We have successfully applied a new structure-based QSAR formalism to study the SAR (Structure-activity Relationship) of 35 PDE-4 inhibitors. This new QSAR formalism describes molecular structures based on the matching of their pharmacophore feature pairs with those (the reference) of the target binding pocket. This reference is derived from the X-ray crystal structures of the target under study. Thus, these descriptors are target-specific and easy to interpret. The PLS (partial least square) method is then employed to reveal the correlation between these target-specific descriptors and the biological activity, and help identify the critical pharmacophore features for PDE-4 inhibition.
To ensure the predictive ability of the built QSAR models, we have adopted a rigorous model validation protocol that emphasizes rational design of the training and test sets, and uses the statistics calculated for both the training set (r2) and test set (R2) as the model selection criteria. Consequently, our models display highly balanced predictive ability for both the training and the test sets (Table 3). In contrast, CoMFA and CoMSIA models have shown signs of over fitting, with high r2 for the training set but much lower R2 for the test set. Thus, this model validation protocol based on both training and test set statistics is critical for the success of this study.
The new structure-based descriptors overcome the drawbacks of traditional descriptors that ignore the binding pocket information. These target-specific descriptors could offer structural insights into the critical features responsible for the potency of the inhibitors. The feature importance analysis proposed in this study has helped fulfill this task. Thus, our models afford both predictive and descriptive models for PDE-4 inhibitors.
Since the structures of many other PDE enzymes have also been solved (e.g., PDE5: 2h44, 2h42, 2h40; PDE-9: 2hd1; PDE3B: 1soj, 1so2; PDE2A: 1z1l and PDE1B: 1taz), the success of this study has also laid a solid foundation for studying the inhibition mechanisms of other PDE enzymes using this new structure-based QSAR formalism. Such a systematic study of the whole family of PDE enzymes will eventually afford insights into the structural requirements for both the potency and selectivity of PDE inhibition, and ultimately contributes to chemical genomics research and therapeutic development targeting the PDE family of enzymes.
ACKNOWLEDGEMENTS
We thank Inte:Ligand for their generous software support with LigandScout. Both X.D. and W.Z. would like to acknowledge the financial support from the Golden Leaf Foundation through the BRITE Institute, NCCU. W.Z. acknowledges grant support from NIH (P20HG003898 and R21GM076059) and the CHDI Foundation.
REFERENCES
| [1] | Torphy TJ. Phosphodiesterase isozymes: molecular targets for novel antiasthma agents Am J Respir Crit Care Med 1998; 157(2): 351-70. |
| [2] | Soderling SH, Beavo JA. Regulation of cAMP and cGMP signaling: new phosphodiesterases and new functions Curr Opin Cell Biol 2000; 12(2): 174-9. |
| [3] | Beavo JA, Conti M, Heaslip RJ. Multiple cyclic nucleotide phosphodiesterases Mol Pharmacol 1994; 46(3): 399-405. |
| [4] | McCullough AR, Steidle CP, Klee B, Tseng LJ. Randomized, double-blind, crossover trial of sildenafil in men with mild to moderate erectile dysfunction: efficacy at 8 and 12 hours postdose Urology 2008; 71(4): 686-92. |
| [5] | Brown WM. Treating COPD with PDE 4 inhibitors Int J Chron Obstruct Pulmon Dis 2007; 2(4): 517-33. |
| [6] | Guazzi M, Samaja M. The role of PDE5-inhibitors in cardiopulmonary disorders: from basic evidence to clinical development Curr Med Chem 2007; 14(20): 2181-91. |
| [7] | Halene TB, Siegel SJ. PDE inhibitors in psychiatry--future options for dementia, depression and schizophrenia? Drug Discov Today 2007; 12(19-20): 870-. |
| [8] | Ghavami A, Hirst WD, Novak TJ. Selective phosphodiesterase (PDE)-4 inhibitors: a novel approach to treating memory deficit? Drugs R D 2006; 7(2): 63-71. |
| [9] | Chen RW, Williams AJ, Liao Z, Yao C, Tortella FC, Dave JR. Broad spectrum neuroprotection profile of phosphodiesterase inhibitors as related to modulation of cell-cycle elements and caspase-3 activation Neurosci Lett 2007; 418(2): 165-9. |
| [10] | Banner KH, Trevethick MA. PDE4 inhibition: a novel approach for the treatment of inflammatory bowel disease Trends Pharmacol Sci 2004; 25(8): 430-6. |
| [11] | Burnouf C, Pruniaux MP. Recent advances in PDE4 inhibitors as immunoregulators and anti-inflammatory drugs Curr Pharm Des 2002; 8(14): 1255-96. |
| [12] | Ducrot P, Andrianjara CR, Wrigglesworth R. CoMFA and CoMSIA 3D-quantitative structure-activity relationship model on benzodiazepine derivatives, inhibitors of phosphodiesterase IV J Comput-Aided Mol Des 2001; 15(9): 767-85. |
| [13] | Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies on thieno[3,2-d]pyrimidines as phosphodiesterase IV inhibitors Bioorg Med Chem Lett 2003; 13(8): 1403-8. |
| [14] | Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. Comparative molecular field analysis (CoMFA) of phthalazine derivatives as phosphodiesterase IV inhibitors Bioorg Med Chem Lett 2003; 13(15): 2473-9. |
| [15] | Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38(11-12): 975-82. |
| [16] | Yang Z, Sun P. 3D-QSAR study of potent inhibitors of phosphodiesterase-4 using a CoMFA approach Int J Mol Sci 2007; 8: 714-22. |
| [17] | Brogle KC, Gund T, Kyle DJ. Improving synthetic efficiency using the computational prediction of biological activity Comb Chem High Throughput Screen 2006; 9(2): 103-3. |
| [18] | Santos-Filho OA, Hopfinger AJ. Structure-based QSAR analysis of a set of 4-hydroxy-5,6-dihydropyrones as inhibitors of HIV-1 protease: an application of the receptor-dependent (RD) 4D-QSAR formalism J Chem Inf Model 2006; 46(1): 345-54. |
| [19] | Romeiro NC, Albuquerque MG, de Alencastro RB, Ravi M, Hopfinger AJ. Free-energy force-field three-dimensional quantitative structure-activity relationship analysis of a set of p38-mitogen activated protein kinase inhibitors J Mol Model 2006; 12(6): 855-68. |
| [20] | Pan D, Tseng Y, Hopfinger AJ. Quantitative structure-based design: formalism and application of receptor-dependent RD-4D-QSAR analysis to a set of glucose analogue inhibitors of glycogen phosphorylase J Chem Inf Comput Sci 2003; 43(5): 1591-607. |
| [21] | Venkatarangan P, Hopfinger AJ. Prediction of ligand-receptor binding thermodynamics by free energy force field three-dimensional quantitative structure-activity relationship analysis: applications to a set of glucose analogue inhibitors of glycogen phosphorylase J Med Chem 1999; 42(12): 2169-79. |
| [22] | Baroni M, Cruciani G, Sciabola S, Perruccio F, Mason JS. A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): theory and application J Chem Inf Model 2007; 47(2): 279-94. |
| [23] | Golbraikh A, Shen M, Xiao Z, Xiao YD, Lee KH, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models J Comput-Aided Mol Des 2003; 17(2-4): 241-53. |
| [24] | Hulme C, Moriarty K, Miller B, et al. The synthesis and biological evaluation of a novel series of indole PDE4 inhibitors I Bioorg Med Chem Lett 1998; 8(14): 1867-72. |
| [25] | Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters J Chem Inf Model 2005; 45(1): 160-9. |
| [26] | Hokuldsson A. A PLS regression methods J Chemometr 1988; 2: 211-8. |
| [27] | Labute P. Quasar-Cluster: A different view of molecular clustering Chem Comp Group J 2002 [Jan 5, 2008]; http://www.chemcomp.com/journal/cluster.htm |