- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Development of Improved Models for Phosphodiesterase-4 Inhibitors with a Multi-Conformational Structure-Based QSAR Method
Adetokunbo Adekoya, Xialan Dong, Jerry Ebalunode, Weifan Zheng*
Abstract
Phosphodiesterase-4 (PDE-4) is an important drug target for several diseases, including COPD (chronic obstructive pulmonary disorder) and neurodegenerative diseases. In this paper, we describe the development of improved QSAR (quantitative structure-activity relationship) models using a novel multi-conformational structure-based pharmacophore key (MC-SBPPK) method. Similar to our previous work, this method calculates molecular descriptors based on the matching of a molecule's pharmacophore features with those of the target binding pocket. Therefore, these descriptors are PDE4-specific, and most relevant to the problem under study. Furthermore, this work expands our previous SBPPK QSAR method by explicitly including multiple conformations of the PDE-4 inhibitors in the regression analysis, and thus addresses the issue of molecular flexibility. The nonlinear regression problem resulted from including multiple conformations has been transformed into a linear equation and solved by an iterative partial least square (iPLS) procedure, according to the Lukacova-Balaz scheme. 35 PDE-4 inhibitors have been analyzed with this new method, and predictive models have been developed. Based on the prediction statistics for both the training set and the test set, these new models are more robust and predictive than those obtained by traditional ligand-based QSAR techniques as well as that obtained with the SBPPK method reported in our previous work. As a result, multiple predictive models have been added to the collection of QSAR models for PDE4 inhibitors. Collectively, these models will be useful for the discovery of new drug candidates targeting the PDE-4 enzyme.
Article Information
Identifiers and Pagination:
Year: 2009Volume: 3
First Page: 54
Last Page: 61
Publisher Id: CCGTM-3-54
DOI: 10.2174/1875397300903010054
Article History:
Received Date: 15/8/2009Revision Received Date: 15/9/2009
Acceptance Date: 17/9/2009
Electronic publication date: 31/12/2009
Collection year: 2009
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.5/), which permits unrestrictive use, distribution, and reproduction in any medium, provided the original work is properly cited.
* Address correspondence to this author at the Department of Pharmaceutical Sciences, BRITE Institute, North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707, USA; Tel: 919 530 6752; Fax: 919 530 6600; E-mail: wzheng@nccu.edu
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 15-8-2009 |
Original Manuscript | Development of Improved Models for Phosphodiesterase-4 Inhibitors with a Multi-Conformational Structure-Based QSAR Method | |
INTRODUCTION
Phosphodiesterases (PDE's) are involved in many cellular signal transductions mediated by cAMP or cGMP molecules. They have been proved to be an important class of drug targets for a variety of diseases. For example, Sildenafil, a PDE-5 inhibitor, has been developed to treat erectile dysfunction (ED) [1McCullough AR, Steidle CP, Klee B, Tseng L. Randomized, double blind, crossover trial of sildenafil in men with mild to moderate erectile dysfunction: efficacy at 8 and 12 hours postdose Urology 2008; 71(4): 686-92.]. Inhibitors of PDE-4 have been studied as potential treatment for COPD (chronic obstructive pulmonary disorder) [2Brown WM. Treating COPD with PDE 4 inhibitors Int J Chron Obstruct Pulmon Dis 2007; 2(4): 517-33., 3Sturton G, Fitzgerald M. Phosphodiesterase 4 Inhibitors for the Treatment of COPD* Chest 2002; 121(5 suppl): 192S-6S.]. Other diseases such as dementia, depression and schizophrenia have also been targeted with PDE inhibitors [4Halene TB, Siegel SJ. PDE inhibitors in psychiatry-future options for dementia, depression and schizophrenia? Drug Discov Today 2007; 12(19-20): 870-.]. Because of the broad biological functions in which PDE enzymes are involved, developing predictive QSAR models for PDE inhibitors may prove to be fruitful for both chemical genomics research and drug discovery targeting the PDE enzymes.
We have been interested in developing predictive QSAR (quantitative structure-activity relationship) models for PDE inhibitors primarily because of their potential role in treating neurodegenerative diseases. For example, selective PDE-4 inhibitors are potential drug candidates for treating memory deficit [5Ghavami A, Hirst WD, Novak TJ. Selective phosphodiesterase (PDE)-4 inhibitors: a novel approach to treating memory deficit? Drugs RD 2006; 7(2): 63-71.] and neurodegeneration [6Chen R, Williams AJ, Liao Z, Yao C, Tortella FC, Dave JR. Broad spectrum neuroprotection profile of phosphodiesterase inhibitors as related to modulation of cell-cycle elements and caspase-3 activation Neurosci Lett 2007; 418(2): 165-9.], and thus were the subject of a previous study by our group [7Dong X, Zheng W. A new structure-based QSAR method affords both descriptive and predictive models for phosphodiesterase-4 inhibitors Curr Chem Genomics 2008; 2(1): 29-39.]. In that study, we have developed a structure-based QSAR model with better predictive power than other published models. However, it has been demonstrated in the literature that multiple models developed with different methodologies tend to be complementary to each other, with each model capturing different aspects of the SAR (structure-activity relationship) trend, and that the joint use of multiple models often enables more effective virtual screening strategy [8Wang R, Wang S. How does consensus scoring work for virtual library screening? An idealized computer experiment J Chem Inf Comput Sci 2001; 41(5): 1422-6., 9Kovatcheva A, Golbraikh A, Oloff S, Xiao Y, Zheng W, Wolschann P, et al. Combinatorial QSAR of ambergris fragrance compounds J Chem Inf Comput Sci 2004; 44(2): 582-95.]. Thus, we aim to develop additional improved models for the PDE-4 inhibitors using a novel QSAR technique.
In our previous work [7Dong X, Zheng W. A new structure-based QSAR method affords both descriptive and predictive models for phosphodiesterase-4 inhibitors Curr Chem Genomics 2008; 2(1): 29-39.], we have demonstrated that the predictiveness of a structure-based QSAR (SB-PPK) model was superior to others that had been developed using more traditional, ligand-based QSAR techniques. This may be due to the fact that the SB-PPK descriptors were generated based on how the inhibitors match the pharmacophore features of the target binding site, and thus they were target-specific; while traditional QSAR methods were ligand-based where no target information was used in calculating the descriptors. Hence, it appeared that target-specific descriptors afforded more predictive models than universal ligand-based descriptors.
One issue that was not addressed in our previous work was that of conformational flexibility, i.e. how to include multiple conformations of an inhibitor in the QSAR analysis. Instead, it allowed only one conformation per inhibitor. In general, this multi-conformational problem has long been an issue in 3D QSAR methodologies. Most current methods, like our previous work, allow only one row of descriptors for each inhibitor in the analysis, and the information of multiple conformations was at best encoded into one row of descriptors. For example, Chen et al. [10Chen X, Rusinko A, Young SS. Recursive partitioning analysis of a large structure activity data set using three-dimensional descriptors1 J Chem Inf Comput Sci 1998; 38(6): 1054-62.] generated multi-feature pharmacophore keys, and features resulted from different conformations were combined into one row of descriptors for a given molecule. The issue with this approach is that after the features from different conformations are encoded into one row of descriptors, it is almost impossible to decode which features come from which conformation. In our current work, we aim to include multiple conformations of each inhibitor in the QSAR analysis to address the issue of conformational flexibility.
We have expanded the Lukacova-Balaz scheme [11Lukacova V, Balaz S. Multimode ligand binding in receptor site modeling: implementation in CoMFA J Chem Inf Comput Sci 2003; 43(6): 2093-105.] for treating multimode issue of 3D QSAR into the SB-PPK methodology, and applied this new multi-conformational QSAR technique to analyze the same set of PDE-4 inhibitors as studied previously for comparative analyses. This new method combines the structure-based QSAR concept (SB-PPK) and a novel mathematical treatment of the multi-conformational issue. We demonstrate that improved models over previous methods can be developed. This multi-conformer approach overcomes the pitfalls of one conformer per ligand paradigm adopted by most traditional 3D QSAR techniques. A standard workflow for model building and validation was employed to ensure the predictiveness of the QSAR models. As a result, all models developed herein give both the training set r2 and the test set R2 over 0.65, indicating a high predictive power. In the following sections, we describe the details for descriptor generation, the theory and the mathematical solution to the multi-conformational issue, followed by results and discussions.
MATERIALS AND METHODS
The PDE-4 Dataset
The 35 indole derivative based PDE-4 inhibitors analyzed by Dong et. al. [7Dong X, Zheng W. A new structure-based QSAR method affords both descriptive and predictive models for phosphodiesterase-4 inhibitors Curr Chem Genomics 2008; 2(1): 29-39.] and Chakraborti et al. [12Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38: 975-82.] were used in this analysis. The dataset covers a diverse set of molecules with a wide range of inhibitory activities against PDE-4. The pIC50 values range from 5.85 to 8.49. The inhibitory activity values are presented in Table 1 and the molecular structures are included in the Supplementary Materials.
The Multi-Conformer QSAR Table
In this work, the input QSAR table for regression analysis is different from a traditional one. Traditional QSAR table has one row for each inhibitor, where the row of descriptors uniquely characterizes the molecule under study. Since multiple conformers are allowed in this multi-conformer QSAR method, multiple rows of descriptors are generated for each inhibitor. We first describe how a single conformer is characterized using the SB-PPK approach, and then discuss the descriptors of multiple conformers for a given inhibitor.
Descriptors for a Single Conformer
The SB-PPK (structure-based pharmacophore key) descriptors for a given conformer of an inhibitor are derived from comparing and matching the pharmacophore feature pairs of the target binding site and the pharmacophore feature pairs of the conformer under study.
As detailed in our previous work [7Dong X, Zheng W. A new structure-based QSAR method affords both descriptive and predictive models for phosphodiesterase-4 inhibitors Curr Chem Genomics 2008; 2(1): 29-39.], the pharmacophore feature pairs of the binding site are calculated by (1) identifying structure-based pharmacophore centers from the target-ligand complex structure, and (2) creating all possible pairs of these feature centers, resulting in feature pair keys. In our previous work, LigandScout [13Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters J Chem Inf Model 2005; 45(1): 160-9.] was employed to find the pharmacophore centers. Here, we employed a different approach to identify the pharmacophore centers. This approach illustrated in Fig. (1 ) uses Delauney tessellation [14Liang J, Edelsbrunner H, Fu P, Sudhakar PV, Subramaniam S. Analytical shape computation of macromolecules: II. Inaccessible cavities in proteins Protein Struct Funct Genet 1998; 33(1): 18-29., 15Liang J, Edelsbrunner H, Fu P, Sudhakar P, Subramaniam S. Analytical shape computation of macromolecules: I. Molecular area and volume through alpha shape Proteins 1998; 33(1): 17-.] of the inhibitor-protein complex structure (1ro6) to identify the interacting atoms of the bound inhibitor and the binding pocket atoms. The space occupied by the Delauney tetrahedra that contain at least one inhibitor atom will be considered the binding pocket space. This space is further approximated by a regular geometric grid with grid spacing of 0.52Å. The grid points that are too close to the protein atoms are trimmed off, leading to a refined geometric grid. The grid points are then labeled based on their distances to nearby protein atoms and the nature of the nearby protein atoms. For example, if a hydrogen bond donor is found in nearby protein atoms, the grid points will be labeled as potential hydrogen bond acceptor. If a positively charged protein atom is found nearby, the grid points are labeled as potentially negatively charged features to complement the protein atom. The whole geometric grid is labeled according to similar principles. Since multiple neighboring grid points are often labeled as the same pharmacophore type, they will be grouped into one pharmacophore center. This clustering is achieved using the ART-2a algorithm [16Carpenter GA, Grossberg S, Rosen DB. Art 2-A: an adaptive resonance algorithm for rapid category learning and recognition Neural Netw 1991; 4(4): 493-504.], a neural network based clustering technique. In this work, 12 pharmacophore centers were generated from the inhibitor-protein complex (1ro6). These pharmacophore centers characterize the unique environment of the binding pocket of the protein. Each center can be one or more of the five pharmacophore types: positively charged (P), negatively charged (N), hydrophobic (H), hydrogen bond donor (D) and hydrogen bond acceptor (A). Pharmacophore feature pairs are then generated by making all possible pairwise combinations of the identified pharmacophore centers. For example, if 5 different pharmacophore centers were found, a total of 10 possible feature pairs could be generated. Each feature pair is determined by the two pharmacophore types involved and the unique distance between the two pharmacophore centers. If the 5 centers were of the following types: P, D, A, H, H, the pharmacophore feature pairs could be PD5, PA7, PH7.5, PH9.0, DA6, DH7, DH5, AH4, AH5, HH3, where the two letters indicate the feature pair key and the number being the distance between the two centers. This set of feature pairs, each having a fixed distance, will be the reference to which the ligand feature pairs are compared in order to generate the SB-PPK descriptors for a given ligand conformer.
) uses Delauney tessellation [14Liang J, Edelsbrunner H, Fu P, Sudhakar PV, Subramaniam S. Analytical shape computation of macromolecules: II. Inaccessible cavities in proteins Protein Struct Funct Genet 1998; 33(1): 18-29., 15Liang J, Edelsbrunner H, Fu P, Sudhakar P, Subramaniam S. Analytical shape computation of macromolecules: I. Molecular area and volume through alpha shape Proteins 1998; 33(1): 17-.] of the inhibitor-protein complex structure (1ro6) to identify the interacting atoms of the bound inhibitor and the binding pocket atoms. The space occupied by the Delauney tetrahedra that contain at least one inhibitor atom will be considered the binding pocket space. This space is further approximated by a regular geometric grid with grid spacing of 0.52Å. The grid points that are too close to the protein atoms are trimmed off, leading to a refined geometric grid. The grid points are then labeled based on their distances to nearby protein atoms and the nature of the nearby protein atoms. For example, if a hydrogen bond donor is found in nearby protein atoms, the grid points will be labeled as potential hydrogen bond acceptor. If a positively charged protein atom is found nearby, the grid points are labeled as potentially negatively charged features to complement the protein atom. The whole geometric grid is labeled according to similar principles. Since multiple neighboring grid points are often labeled as the same pharmacophore type, they will be grouped into one pharmacophore center. This clustering is achieved using the ART-2a algorithm [16Carpenter GA, Grossberg S, Rosen DB. Art 2-A: an adaptive resonance algorithm for rapid category learning and recognition Neural Netw 1991; 4(4): 493-504.], a neural network based clustering technique. In this work, 12 pharmacophore centers were generated from the inhibitor-protein complex (1ro6). These pharmacophore centers characterize the unique environment of the binding pocket of the protein. Each center can be one or more of the five pharmacophore types: positively charged (P), negatively charged (N), hydrophobic (H), hydrogen bond donor (D) and hydrogen bond acceptor (A). Pharmacophore feature pairs are then generated by making all possible pairwise combinations of the identified pharmacophore centers. For example, if 5 different pharmacophore centers were found, a total of 10 possible feature pairs could be generated. Each feature pair is determined by the two pharmacophore types involved and the unique distance between the two pharmacophore centers. If the 5 centers were of the following types: P, D, A, H, H, the pharmacophore feature pairs could be PD5, PA7, PH7.5, PH9.0, DA6, DH7, DH5, AH4, AH5, HH3, where the two letters indicate the feature pair key and the number being the distance between the two centers. This set of feature pairs, each having a fixed distance, will be the reference to which the ligand feature pairs are compared in order to generate the SB-PPK descriptors for a given ligand conformer.
 |
Fig. (1) Flowchart for the generation of receptor pharmacophore feature pairs. |
The pharmacophore feature pairs for a given conformer of a ligand can be generated according to our previous work [7Dong X, Zheng W. A new structure-based QSAR method affords both descriptive and predictive models for phosphodiesterase-4 inhibitors Curr Chem Genomics 2008; 2(1): 29-39.] and outlined in Fig. (2 ). Instead of using the LigandScout program, the atom typing program Patty (OE Scientific, NM, USA) was employed in this work to label each atom of an inhibitor molecule. Similar pharmacophore types are defined: positively charged (P), negatively charged (N), hydrophobic (H), hydrogen bond donor (D) and hydrogen bond acceptor (A). Once each atom is typed with a proper pharmacophore type, pharmacophore feature pairs can be generated in a way similar to how the reference (target binding site) pharmacophore feature pairs are generated. If a conformer had the following pharmacophore centers: A, P, A, D, then 6 possible pharmacophore feature pairs could be generated: AP6.9, AA5, AD6, PA9, PD4.8, AD7, where the two letters indicate the feature pair key and the number being the distance between the two feature centers.
). Instead of using the LigandScout program, the atom typing program Patty (OE Scientific, NM, USA) was employed in this work to label each atom of an inhibitor molecule. Similar pharmacophore types are defined: positively charged (P), negatively charged (N), hydrophobic (H), hydrogen bond donor (D) and hydrogen bond acceptor (A). Once each atom is typed with a proper pharmacophore type, pharmacophore feature pairs can be generated in a way similar to how the reference (target binding site) pharmacophore feature pairs are generated. If a conformer had the following pharmacophore centers: A, P, A, D, then 6 possible pharmacophore feature pairs could be generated: AP6.9, AA5, AD6, PA9, PD4.8, AD7, where the two letters indicate the feature pair key and the number being the distance between the two feature centers.
 |
Fig. (2) Flowchart for ligand pharmacophore key generation. |
After both the reference feature pairs and the ligand feature pairs are generated, the SB-PPK descriptors for a given ligand conformer are generated based on the matching of the feature pairs of the conformer with the feature pairs of the binding pocket (i.e. the reference). The value of a given descriptor is the total number of matches for that descriptor between the ligand and the binding pocket. More details can be found in our previous publication [7Dong X, Zheng W. A new structure-based QSAR method affords both descriptive and predictive models for phosphodiesterase-4 inhibitors Curr Chem Genomics 2008; 2(1): 29-39.].
Descriptors for Multiple Conformers of an Inhibitor.
Multiple conformers were generated for each inhibitor using the OMEGA program, version 2.0 (OMEGA, OE Scientific, NM, USA). The maximum number of conformers for each molecule was set to 1000. Top ranking conformers were retained for each inhibitor, and each conformer was used to generate its SB-PPK descriptors as described above. As a result, multiple rows of SB-PPK descriptors were generated for each inhibitor. For example, if 10 conformers were used for each molecule, 350 rows of descriptors would be generated for a dataset of 35 molecules. Table 2 shows an example of the multi-conformer QSAR table.
The above multi-conformer QSAR table will be used as the input for the iterative partial least square (iPLS) procedure to derive multi-conformational QSAR models, according to the Lukacova-Balaz scheme [11Lukacova V, Balaz S. Multimode ligand binding in receptor site modeling: implementation in CoMFA J Chem Inf Comput Sci 2003; 43(6): 2093-105.].
Generation of Multi-Conformational QSAR Models Using the Lukacova-Balaz Scheme
Based on a similar set of equations as those employed by Lukacova & Balaz [11Lukacova V, Balaz S. Multimode ligand binding in receptor site modeling: implementation in CoMFA J Chem Inf Comput Sci 2003; 43(6): 2093-105.], the multi-conformational QSAR models are built using the iPLS method. One major difference between our work and that of Lukacova lies in how the structure-based descriptors are generated. The details of the theory are discussed in the context of the SB-PPK descriptors as follows.
Let Ki, the total dissociation constant of a ligand bound to a receptor in multiple conformations be expressed as:
Where kij is the partial dissociation constant, i is the ligand and j is a conformer of ligand i.
Similar to Lukacova et. al., we propose a correlation between the partial dissociation constant (kij) and the structure-based descriptors nijh, such that:
Where H is the number of descriptors, co is the intercept and ch is the regression coefficient.
Substituting Eq. (2) into Eq. (1), we have
From Eq. (3), we can develop a structure-based multi-conformational QSAR model by solving for co and ch since we know Ki and nijh for each ligand. Eq. (3), a non-linear equation, has been transformed into a linear one, according to Lukacova et. al. [11Lukacova V, Balaz S. Multimode ligand binding in receptor site modeling: implementation in CoMFA J Chem Inf Comput Sci 2003; 43(6): 2093-105.]. The difference between our linear equation shown in Eq. (4) and that of Lukacova's is the summation of the kij, which are the denominators in our work as opposed to numerators in Lukacova's. We find this to be a much better fit for the iterative PLS method.
To solve the above equation for the coefficients, i. e. building a multi-conformational QSAR model, an iterative PLS procedure was employed. The left of equation 4 is the new dependent variable for the PLS analysis; and the independent variables for the iPLS procedure are not the original descriptors, nijh; rather, they are functions of partial association constants (kij) and the original descriptors (nijh), as shown in equation 4. In fact, the PLS variables also depend on the regression coefficients ch (h=1, 2, . . H) in each iteration. Once the regression coefficients are set in the first iteration, new kij values are calculated from equation 2, and then used to update the variables of equation 4. This procedure is repeated until the resultant models converge. The initial PLS variables for equation 4 are set as the average values of descriptors over all the conformations for each of the inhibitors. Once the iPLS procedure converges, the final coefficients (co to ch) define a QSAR model, and equation 3 is used to predict the activity values for unknown or test set molecules from their multi-conformational descriptors (nijh).
Integrated Workflow for Model Building and Validation
We have adopted a standard workflow reported previously [17Golbraikh A, Shen M, Xiao Z, Xiao Y, Lee K, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models J Comput Aided Mol Des 2003; 17(2-4): 241-53.] to train and validate QSAR models. As shown in Fig. (3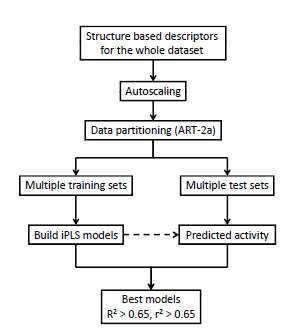 ), the splitting of the dataset into training and test sets as well as obtaining the statistics for both the training set and test set is the key in this workflow. A given dataset is first divided into multiple pairs of training and test sets by a clustering procedure based on the ART-2a algorithm [16Carpenter GA, Grossberg S, Rosen DB. Art 2-A: an adaptive resonance algorithm for rapid category learning and recognition Neural Netw 1991; 4(4): 493-504.]. The parameters in ART-2a algorithm were adjusted in order to obtain 7 multi-member clusters, and one molecule from each cluster was randomly selected into a test set, resulting in multiple 7-member test sets. Once molecules were selected into a test set, the remaining molecules in the dataset were put into the corresponding training set. In this work, 30 pairs of training and test sets were generated, with the training set having 28 molecules and the test sets having 7 molecules. For each pair of training and test sets, the iPLS procedure was used to train the models on the training set, and the developed models were used to predict the 7 test set molecules. Model quality was calculated as r2 for the training set, and R2 for the test set. While r2 reflects the model quality on the training set, R2 indicates the predictive power of the model against the test set. Only models with both indicators (r2 and R2) greater than a preset threshold (0.65 in this work) were retained as the final models for future use. The use of 0.65 as the cutoff value is arbitrary. One can use higher values as well. If no good models can be found with this cutoff, one can lower it. The ultimate goal is to find models that afford both r2 and R2 greater than a preset threshold.
), the splitting of the dataset into training and test sets as well as obtaining the statistics for both the training set and test set is the key in this workflow. A given dataset is first divided into multiple pairs of training and test sets by a clustering procedure based on the ART-2a algorithm [16Carpenter GA, Grossberg S, Rosen DB. Art 2-A: an adaptive resonance algorithm for rapid category learning and recognition Neural Netw 1991; 4(4): 493-504.]. The parameters in ART-2a algorithm were adjusted in order to obtain 7 multi-member clusters, and one molecule from each cluster was randomly selected into a test set, resulting in multiple 7-member test sets. Once molecules were selected into a test set, the remaining molecules in the dataset were put into the corresponding training set. In this work, 30 pairs of training and test sets were generated, with the training set having 28 molecules and the test sets having 7 molecules. For each pair of training and test sets, the iPLS procedure was used to train the models on the training set, and the developed models were used to predict the 7 test set molecules. Model quality was calculated as r2 for the training set, and R2 for the test set. While r2 reflects the model quality on the training set, R2 indicates the predictive power of the model against the test set. Only models with both indicators (r2 and R2) greater than a preset threshold (0.65 in this work) were retained as the final models for future use. The use of 0.65 as the cutoff value is arbitrary. One can use higher values as well. If no good models can be found with this cutoff, one can lower it. The ultimate goal is to find models that afford both r2 and R2 greater than a preset threshold.
 |
Fig. (3) Overall workflow of structure-based QSAR method. |
RESULTS AND DISCUSSIONS
Fast Convergence of the Iterative PLS Analysis
As shown in Fig. (4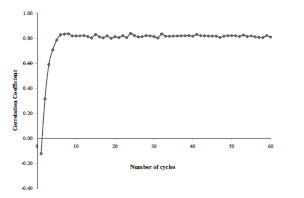 ), the iPLS procedure to obtain the multi-conformational QSAR model converges very fast. In just 7 cycles of PLS regression, the models reach r2 of about 0.83, and maintain at that level. This procedure is highly efficient compared to other approaches, where all combinations of conformers need to be considered to build QSAR models, and stochastic optimization process is often used to select the best performing models. Because many conformers are allowed to represent an inhibitor molecule, conformational flexibility is taken into account in our QSAR models. This is better than traditional single conformer based 3D QSAR techniques, which is demonstrated in the following sections.
), the iPLS procedure to obtain the multi-conformational QSAR model converges very fast. In just 7 cycles of PLS regression, the models reach r2 of about 0.83, and maintain at that level. This procedure is highly efficient compared to other approaches, where all combinations of conformers need to be considered to build QSAR models, and stochastic optimization process is often used to select the best performing models. Because many conformers are allowed to represent an inhibitor molecule, conformational flexibility is taken into account in our QSAR models. This is better than traditional single conformer based 3D QSAR techniques, which is demonstrated in the following sections.
 |
Fig. (4) Fast convergence of the iterative PLS analysis. |
Effect of Number of Principal Components on Model Quality
We used different numbers of principal components to obtain the PLS QSAR models, the results of which are shown in Fig. (5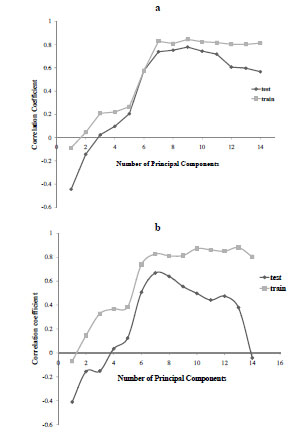 ). These results are indicative of the predictiveness of the QSAR models. The r2 values of the training set and the R2 values of the test set are shown on the y-axis and the number of principal components on the x-axis. In the first case, the r2 values increases monotonically with increase in the number of principal components for the training set. This trend is also observed in the R2 values for the test set, however, they reach a plateau at around 7 principal components, and starts to decrease in value from 10 principal components onward. This decrease in the R2 values indicated over-training when too many principal components were used in the PLS model. In the second case, R2 values decreased much faster than in the first case. The models with number of principal components between 6 and 9 were predictive for both the training and test sets. Based on this observation, we have chosen to build models using 7 principal components to derive both model-1 and model-2 reported below.
). These results are indicative of the predictiveness of the QSAR models. The r2 values of the training set and the R2 values of the test set are shown on the y-axis and the number of principal components on the x-axis. In the first case, the r2 values increases monotonically with increase in the number of principal components for the training set. This trend is also observed in the R2 values for the test set, however, they reach a plateau at around 7 principal components, and starts to decrease in value from 10 principal components onward. This decrease in the R2 values indicated over-training when too many principal components were used in the PLS model. In the second case, R2 values decreased much faster than in the first case. The models with number of principal components between 6 and 9 were predictive for both the training and test sets. Based on this observation, we have chosen to build models using 7 principal components to derive both model-1 and model-2 reported below.
 |
Fig. (5) The effect of number of principal components on model quality for model-1 (top) and model-2 (bottom). |
Effect of Number of Conformers per Inhibitor on Model Quality
It is essential when building a multi-conformer QSAR model to find out the optimal number of conformers to use. We have tested a variety of number of conformers in this work in order to find out the best conformer number. When 5 conformers were used, R2 values were low, with only 1 in 100 models above 0.50. On the other hand, when 10 conformers were used, the values of the R2 above 0.5 were around 6 in 100. Further experimentation led us to conclude that 7 conformers for each ligand afforded the best models. In the rest of this study, we present the results obtained with 7 conformers per inhibitor in the models.
Effect of Dataset Splitting on Model Quality
The results presented in this work are based on a rational splitting of the dataset into training and test sets. We found that by introducing a more rational method, there is a higher occurrence of models with R2 higher than 0.5. A comparison of the results between rational splitting and random splitting showed that out of 20 splits, 9 gave models (7 conformers and 7 principal components) with R2 values above 0.5 for the rational splitting, compared to only 1 model having R2 above 0.5 based on random splitting. The R2 value in the random split was 0.74 while the R2 values for the rational splitting were 0.54, 0.74, 0.68, 0.71, 0.64, 0.67, 0.69, 0.56 and 0.74. A fewer number of splitting cycles is required to obtain a predictive model. This observation is consistent with that of Golbraikh et al. [17Golbraikh A, Shen M, Xiao Z, Xiao Y, Lee K, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models J Comput Aided Mol Des 2003; 17(2-4): 241-53.]. Thus, all models were built using the training and test sets created with the clustering-based splitting of the dataset.
Predictiveness of MC-SBPPK Models Compared to Previous Methods
The predictive power of the new models developed with the MC SBPPK method exceeds that of other models developed with more traditional QSAR techniques [12Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies of indole derivatives as phosphodiesterase IV inhibitors Eur J Med Chem 2003; 38: 975-82., 18Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. 3D-QSAR studies on thieno[3,2-d]pyrimidines as phosphodiesterase IV inhibitors Bioorg Med Chem Lett 2003; 13(8): 1403-8., 19Chakraborti AK, Gopalakrishnan B, Sobhia ME, Malde A. Comparative molecular field analysis (CoMFA) of phthalazine derivatives as phosphodiesterase IV inhibitors Bioorg Med Chem Lett 2003; 13(15): 2473-9.], and that of the model developed with our previous SB-PPK method [7Dong X, Zheng W. A new structure-based QSAR method affords both descriptive and predictive models for phosphodiesterase-4 inhibitors Curr Chem Genomics 2008; 2(1): 29-39.]. For example, a MOE (Chemical Computing Group, Montreal, Canada) based 2D QSAR method afforded models with r2 of 0.66 for the training set, and R2 of 0.58 for the test set [7Dong X, Zheng W. A new structure-based QSAR method affords both descriptive and predictive models for phosphodiesterase-4 inhibitors Curr Chem Genomics 2008; 2(1): 29-39.]. A reported CoMFA model gave an r2 of 0.986, but an R2 of 0.560, indicating overtraining on the training set and underperforming on the test set. A CoMSIA model gave an r2 of 0.967, but an R2 of 0.590 for the test set, again indicating overtraining and underperforming during prediction. The most balanced model developed in our previous work afforded an r2 of 0.75 for the training and R2 of 0.624 for the test set. In the current work, the two best models gave r2 of 0.83 and 0.83 for the training sets, and R2 of 0.74 and 0.67 for the test sets. Figs. (6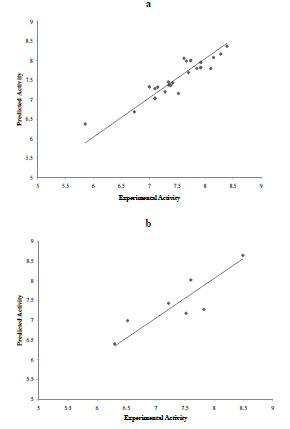 , 7
, 7 ) show the scatter plots of predicted vs. experimental activities. Figs. (8
) show the scatter plots of predicted vs. experimental activities. Figs. (8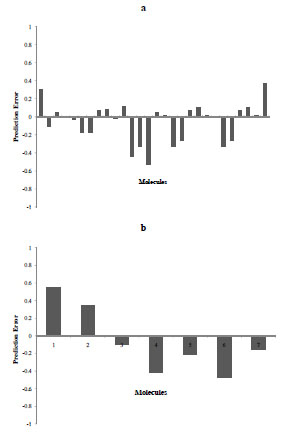 , 9
, 9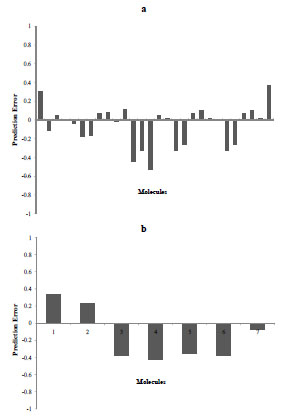 ) show the absolute errors of prediction for both MC SBPPK models. Thus, the MC SBPPK method has successfully incorporated multi-conformers in the QSAR analysis, and afforded more predictive models than all previously reported methods.
) show the absolute errors of prediction for both MC SBPPK models. Thus, the MC SBPPK method has successfully incorporated multi-conformers in the QSAR analysis, and afforded more predictive models than all previously reported methods.
 |
Fig. (6) Predicted activity vs. experimental activity for the training set (top) and test set (bottom) for model 1. |
 |
Fig. (7) Predicted activity vs. experimental activity for the training set (top) and test set (bottom) for model 2. |
 |
Fig. (8) Prediction error of training set (top) and test set (bottom) for model 1. |
 |
Fig. (9) Prediction error of training set (top) and test set (bottom) for model 2. |
CONCLUSIONS
We described the development of improved QSAR models based on a new multi-conformational structure-based pharmacophore key (MC SBPPK) method. Following our previous work, this method calculates molecular descriptors based on the matching of a molecule's pharmacophore features with those of the target binding pocket. Therefore, these pharmacophore key (SBPPK) descriptors are PDE4-specific, and most relevant to the problem under study. We also successfully expanded our previous SBPPK method by explicitly including multiple conformations of PDE-4 inhibitors in the analysis, and thus addresses the issue of molecular flexibility. The nonlinear regression problem resulted from including multiple conformations has been transformed into a linear equation and solved by an iterative partial least square (iPLS) procedure, according to the Lukacova-Balaz scheme originally developed to address the multi-mode issue in 3D QSAR analysis. We demonstrated that more predictive models were obtained by the MC SBPPK method, indicating the importance of incorporating multiple conformations to address the issue of molecular flexibility in 3D QSAR modeling.
The success of the MC SBPPK method in modeling the PDE-4 inhibitors also helps to establish the platform of structure-based multi-conformational QSAR method for modeling inhibitors of other PDE enzymes. The fact that many X-ray structures of PDE enzymes have been solved supports the use of structure-based QSAR methods. Ultimately, we will develop a collection of MC SBPPK models for PDE inhibitors, and contribute to the chemical genomics research of the PDE family of enzymes.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Web site along with the published article Download Supplementary Material.
ACKNOWLEDGEMENTS
We would like to acknowledge the financial support by the Golden Leaf Foundation through the BRITE Center, North Carolina Central University. We also acknowlegde the generous software support from OpenEye Scientific Software. W.Z. acknowledges funding (1SC3GM086265) from NIH (National Institutes of Health).