
The Open Arthritis Journal
(Discontinued)
ISSN: 1876-5394 ― Volume 7, 2014
Design of the Rituximab in ANCA-Associated Vasculitis (RAVE) Trial
Ulrich Specks, Peter A Merkel, Gary S Hoffman, Carol A Langford, Robert Spiera, Phil Seo, Cees G.M Kallenberg, E. William St Clair, Linna Ding, Lisa Webber, Masoud Mokhtarani, Nadia K Tchao, Peter Sayre, Vicky Seyfert-Margolis, David Ikle, Paul Brunetta P, David Zhang, Lourdes Sejismundo, Mark Mueller, John H Stone*Author Comment: for the RAVE-ITN Research Group#
Abstract
Granulomatosis with polyangiitis (formerly Wegener's) (GPA) and microscopic polyangiitis (MPA) share many clinical and pathological features, including antineutrophil cytoplasmic antibodies (ANCA) directed against either proteinase 3 (PR3) or myeloperoxidase (MPO). These two “ANCA-associated” vasculitides (AAV) are associated with a high mortality in untreated patients, substantial morbidity from standard therapies, and a significant risk of disease relapse.
The Rituximab in ANCA-Associated Vasculitis (RAVE) trial is a randomized, double-blind, double-dummy, active-controlled, non-inferiority trial of a new approach to the induction of remission. The RAVE trial represents the first challenge of a biologic agent to CYC as the standard of care for remission induction in AAV. The primary outcome analysis, reported in 2010, compared conventional therapy (the combination of cyclophosphamide (CYC) and glucocorticoids) to the combination of rituximab (RTX) and glucocorticoids. Longer term outcomes to 18 months and beyond have not been reported.
The trial aimed to determine if the combination of RTX plus glucocorticoids was non-inferior to the combination of CYC and glucocorticoids. To test this hypothesis, eight clinical centers planned to enroll 200 patients. The randomization was stratified by center and by ANCA subtype. Patients were assigned randomly to each treatment arm in an allocation ratio of 1:1. The primary outcome had two components: 1) the ability of the assigned regimen to induce disease remission by month 6; and, 2) successful discontinuation of prednisone by month 6. All primary analyses were performed on an intention-to-treat basis. A major secondary outcome of interest was the restoration of immune tolerance, defined as disease quiescence and the absence of ANCA following the reconstitution of normal B cell numbers. To meet this definition, patients were required to achieve and maintain disease remissions, complete the prednisone taper, and remain on no immunosuppressive medications after discontinuing prednisone. Patients were followed for 18 months after the final patient was enrolled to evaluate the impact of the two treatment regimens on tolerance restoration.
In this paper, we describe the development and design of the RAVE trial as a pivotal trial in an orphan disease indication. We illuminate the unique challenges involved in comparing a new treatment approach against an entrenched standard of care in a double-blind, double-dummy trial of a biologic for the treatment of a rare disease.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 4
First Page: 1
Last Page: 18
Publisher Id: TOARTHJ-4-1
DOI: 10.2174/1876539401104010001
Article History:
Received Date: 15/7/2011Revision Received Date: 30/8/2011
Acceptance Date: 22/9/2011
Electronic publication date: 16/11/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Rheumatology Unit, Massachusetts General Hospital, 55 Fruit St./Yawkey 2C, Boston, MA. 02114, USA: Tel: 617-643-1274; Fax: 617-643-1274; E-mail: jhstone@partners.org* ClinicalTrials.gov identifier: NCT00104299.
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 15-7-2011 |
Original Manuscript | Design of the Rituximab in ANCA-Associated Vasculitis (RAVE) Trial | |
1. INTRODUCTION
Granulomatosis with polyangiitis (formerly Wegener's) (GPA) and microscopic polyangiitis (MPA) are two closely-related forms of systemic vasculitis that affect small- to medium-sized blood vessels [1]. Because most patients with GPA and MPA have circulating anti-neutrophil cytoplasmic antibodies (ANCA), they are often termed “ANCA-associated vasculitides” (AAV) [2-4]. Untreated severe AAV is fatal [5]. For the last 40 years, standard therapy has consisted of glucocorticoids combined with cyclophosphamide (CYC). Although this treatment regimen is successful in inducing at least temporary remissions in the majority of patients [6-10], the management of patients with AAV remains challenging. Long-term CYC use is associated with well-recognized toxicities [7,8,11]. For this reason, long-term use of CYC has been replaced by staged regimens in which CYC is replaced by methotrexate or azathioprine (AZA) after 3-6 months, once clinical remission is achieved [9,10,12-14]. However, repeated and long-term exposure to CYC remains a safety concern because of the high disease relapse rate [10,15,16]. To date, the focus of randomized controlled trials in severe AAV has been to minimize the duration and cumulative dose of CYC rather than to identify an agent with greater or equal efficacy that can replace for remission induction [9,10,17].
Rituximab (RTX), a chimeric monoclonal anti-CD20 antibody that targets B lymphocytes specifically, is a potential replacement for CYC in AAV [18]. CYC has a profound effect on B lymphocytes in AAV, which may in part explain its efficacy for the treatment of this disease [19-21]. Moreover, preliminary results with RTX therapy in patients with refractory disease suggest that this agent was both effective and well tolerated in AAV [22-24]. Given the B cell depleting effects of RTX, we designed a large clinical trial of this agent in AAV to determine the extent to which B cells play a role in the pathogenesis of this disease, incorporating mechanistic studies to explore potential treatment pathways.
Attempts to design and conduct of randomized controlled trials in rare diseases such as AAV confront multiple challenges [25]. First, severe, untreated AAV is fatal, but the existing standard of care is effective (albeit toxic). Given the efficacy of CYC, a placebo-controlled trial with a new agent would have been unethical [26]. (The established standard of care for remission induction (CYC) has never been compared to placebo). Second, because of the perceived efficacy of CYC in inducing remission, a superiority trial was felt impractical because of sample size considerations. For these two reasons, we compared RTX and CYC therapy head-to-head using a non-inferiority approach, relying upon the historical experience of untreated controls to support the case for superiority of RTX over placebo.
The orphan disease status posed an additional challenge to designing this trial. Patients with severe AAV require expert multidisciplinary specialty care, which is concentrated within only a few specialized centers. To ensure timely enrollment, a multicenter effort involving as many of these centers as possible was necessary. Finally, although the design required that only patients with AAV for whom CYC was considered the standard of care would be eligible, we felt it was highly desirable to look for consistency of treatment effects across meaningful clinical subsets of subjects.
With these considerations in mind, we designed the head-to-head comparison of RTX to CYC as a non-inferiority trial with the intention of providing persuasive, internally-consistent evidence of the efficacy of RTX for both primary and secondary endpoints. We intended this study to serve as a pivotal trial of RTX in AAV and for its results to constitute sufficient support for an effectiveness claim to the United States Food & Drug Administration (FDA). Herein, we describe the rationale, design, statistical analysis plan, and conduct of this trial. The primary results of this clinical trial have been published elsewhere (NEJM reference).
2. DESIGN AND METHODS
2.1. Funding
RAVE was supported by the Immune Tolerance Network (ITN), which is funded in turn by the National Institute of Allergy and Infectious Disease (NIAID). RTX, matching placebo, and partial funding were provided by Genentech, Inc. (South San Francisco, CA) and Biogen IDEC, Inc. (Cambridge, MA).
2.2. Organization
The RAVE Research Group comprised eight clinical centers*, a Coordinating Center [Pharmaceutical Products Development, Inc. (PPDI) until April 2009, then Rho,Inc.], a drug distribution center (Eminent Services, Inc., Frederick, MD), and the Tolerance Assay and Data Analysis Group of the ITN (See Mechanistic Studies, below). The list of personnel at the trial sites and all branches of the RAVE Research Group are shown in the Appendix.
Each RAVE clinical site included a primary patient assessor (termed the “Investigator”), a Safety Officer(s), a Trial Coordinator(s), and a laboratory technician. An individual assigned to be the Investigator or the Safety Officer for one patient could fulfill the other role for other patients.
2.3. Trial Objective and Hypothesis
RAVE compared a new treatment (RTX) to a conventional standard of care (CYC), which was first employed in AAV in 1954 [27]. The hypothesis of the trial was that the combination of RTX plus glucocorticoids was not inferior to conventional therapy in its ability to induce disease remission and permit glucocorticoid discontinuation by 6 months. Critical RAVE Trial definitions are shown in Table 1.

2.4. Entry Criteria
Two of the required entry criteria for RAVE were: 1) a clinical diagnosis of GPA or MPA that fulfilled the Chapel Hill Consensus Conference definitions of GPA or MPA [1]; and, 2) presence of PR3-ANCA or MPO-ANCA at trial entry.
2.5. Eligibility Requirements
The eligibility criteria for the trial are summarized in Table 2. All patients enrolled had active disease within 28 days before enrollment, defined by a Birmingham Vasculitis Activity Score for WG (BVAS/WG) of > 3 [28] (see Appendix). Patients who were starting their first courses of treatment for AAV or those suffering a flare of previously quiescent disease were eligible. All patients had severe disease at screening, meaning that outside the trial they would have been treated with CYC and high-dose glucocorticoids.

Patients treated with certain immunosuppressive or biologic agents within specific periods of time before trial entry were ineligible. The reasons for this were two-fold: to prevent confounding of the primary outcome by pre-trial therapy; and to permit certain mechanistic studies on baseline samples (Table 2).
2.6. Assignment to Study Groups
Treatment assignments were generated in permuted blocks. The randomization schedule, generated, written, and controlled by the Coordinating Center, was yielded an assignment ratio of 1:1 between the two treatment groups and within each stratum. The randomization was stratified by both ANCA subtype (PR3- or MPO-ANCA) and clinical site.
2.7. Study Visits
Data collected at the baseline visit are shown in Table 3. At each trial visit, serum, plasma, and peripheral blood mononuclear cell samples were collected. DNA samples were collected at the 2 month visit only (Table 3).

2.8. Trial Phases
There were three treatment phases (Fig.1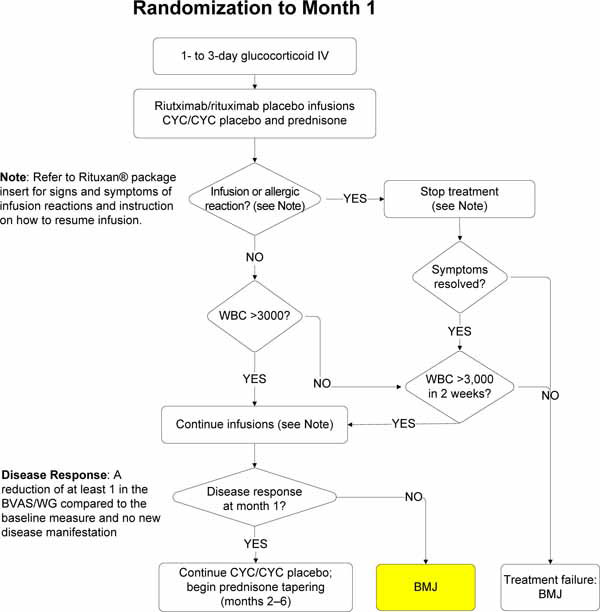 of appendix):
of appendix):
2.8.1. Remission Induction (Randomization through Month 6)
The remission induction phase was defined as the time from the first administration of study treatment through month 6. The primary outcome was assessed at month 6.
2.8.2. Remission Maintenance (Month 6 through Month 18)
The remission maintenance phase began at month 6 and extended through month 18. Data on treatment efficacy and safety obtained during this period will be analyzed for secondary endpoints, including the restoration of immune tolerance. Because all patients will be treated with protocolized medication regimens up to 18 months, efficacy analyses beyond the remission induction phase will focus upon this period.
2.8.3. Best Medical Judgment (After 18 Months)
Following the 18 month visit, all patients discontinued trial therapies and were treated according to best medical judgment. Blinding to the originally assigned treatment was maintained during this phase.
2.8.4. Common Closeout Date
Clinical data collection ended on a common closeout date 18 months after enrollment of the last patient. The eighteen month period was selected because it is regarded as the minimum period of therapy in regimens involving conventional immunosuppressive agents [9]. Thus, patients enrolled early in the trial had significantly longer blinded follow-up periods. The mean duration of follow-up was 29 months (standard deviation [SD] = 13).
2.9. Treatments
2.9.1. Glucocorticoids
Patients in both treatment arms received glucocorticoids. All patients received at least a one 1000 milligram pulse of intravenous methylprednisolone at baseline. Methylprednisolone could be repeated up to a total of 3 doses at the discretion of the investigator. Daily prednisone therapy began the day after completion of the methylprednisolone doses. The starting dose of prednisone was 1 mg/kg, not to exceed 80 mg/day.
The prednisone taper was designed such that before month 6, all patients who had achieved clinical remission and not flared were no longer taking any glucocorticoids. The entire tapering process required 5 months. The protocol mandated the reduction of prednisone to 40 mg/day by the timepoint 1 month after the first RTX infusion. However, at the investigator’s discretion, the prednisone dose could be reduced to 40 mg/d earlier. Once a patient’s dose had been decreased to 40 mg/day and maintained for two weeks, step-wise dose reductions continued every 2 weeks, through daily dose levels of 30 mg, 20 mg, 15 mg, 10 mg, 7.5 mg, 5 mg, 2.5 mg, and 0 mg.
2.9.2. Remission Induction Medications
Patients in the investigational therapy arm received RTX (375 mg/m2 intravenously each week for four doses), daily oral CYC placebo, and glucocorticoids.
Patients in the standard therapy arm received four weekly infusions of RTX placebo, daily CYC (2 mg/kg, with doses modified for renal dysfunction), and glucocorticoids. Patients who achieved disease remissions between three and six months (inclusive) could be switched to the remission maintenance medication – azathioprine (AZA) for the patients randomized to CYC or AZA placebo for those receiving RTX – at that time. However, all patients received a minimum of 3 months of CYC/CYC placebo.
The RTX regimen employed in RAVE was the non-Hodgkin’s lymphoma regimen because it was in AAV preliminary studies [23,24].
2.9.3. Remission Maintenance Medications
Patients in the RTX arm switched from CYC placebo to AZA placebo. Those in the standard therapy arm switched from CYC to AZA (2 mg/kg/day) [9]. The implications of this are critical in evaluating the effects of RTX beyond six months because after the achievement of disease remission and successful completion of the glucocorticoid taper, patients in the RTX were on no active therapy.
2.9.4. Concomitant Medications
All patients received Pneumocystis jiroveci prophylaxis, consisting of daily single-strength trimethoprim/sulfametho-xazole, 80mg/400mg. Patients allergic to sulfa medications were prescribed appropriate alternatives.
2.10. Disease Response
Disease response, assessed formally at month 1, was defined as: 1) no new disease manifestation; 2) no worsening of existing disease; and, 3) reduction of at least 1 in the BVAS/WG compared to the score at screening (See Assessment of DiseaseActivity, below). Early treatment failures were defined as failure to achieve a disease response by month 1. Because this was a blinded trial of a new, unproven therapy compared to an entrenched standard of care regimen that is widely regarded as being effective, the investigators believed it important to ensure that patients were responding to their assigned treatment one month into therapy.
2.11. Treatment Failure
Clinical events that lead to categorization of subjects having experienced a treatment failure (early or otherwise) are shown in Table 4.

2.12. Crossovers
Subjects who experienced severe disease flares between the month 1 and month 6 study visits were crossed over to the opposite treatment arm. This feature was deemed essential to providing equipoise and maintenance of the double-blind design. Crossover patients were considered treatment failures.
2.13. Treatment of Disease Flares
The treatment of disease recurrences was dictated by flare severity and the study phase in which it occurred. An algorithm for the treatment of disease flares is shown in Fig. (2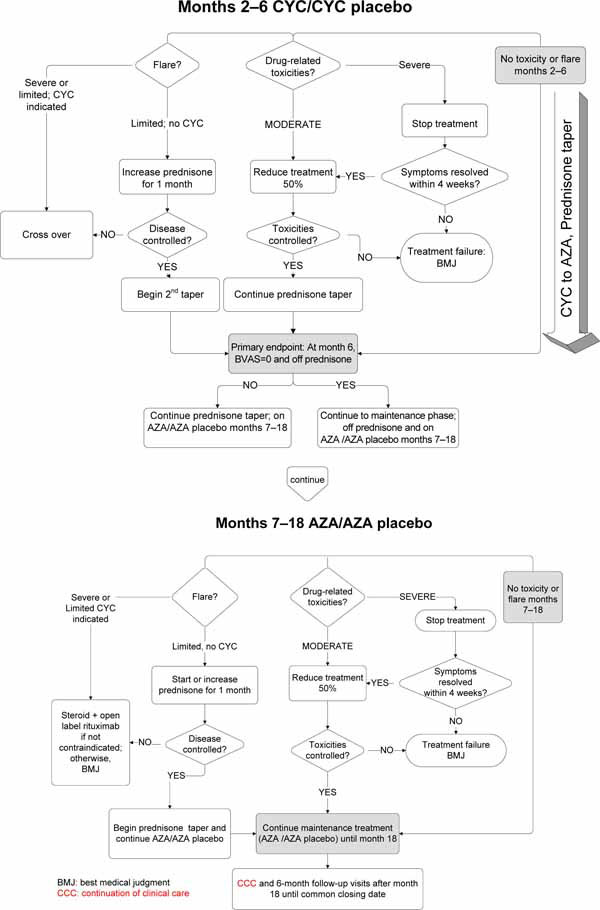 ) (Appendix).
) (Appendix).
2.13.1. Limited Flares
During all trial phases, limited flares were treated by increasing prednisone to a dose selected by the Investigator. The new prednisone dose was maintained for 1 month before resumption of the taper specified in the protocol.
2.13.2. Severe Flares During Remission Induction
Patients with severe flares that occurred during remission maintenance but before month 6 were given the option of crossing over blindly to the other treatment arm. Patients who crossed over to the other treatment arm underwent the full remission induction regimen again, including between one and three 1000 milligram pulses of methylprednisolone.
2.13.3. Severe Flares During Remission Maintenance
Patients with severe flares that occurred between months 6 and 18 were given the option of treatment with open-label RTX, accompanied by the glucocorticoid regimen for remission induction.
2.13.4. Severe Flares During Best Medical Judgment
Severe flares that occurred after 18 months were treated at the discretion of the physician-investigator with either RTX or CYC, both accompanied by pulse methylprednisolone followed by prednisone. These treatments were administered on an open-label basis.
2.14. Data Collection
The types and frequency of data collected for the purpose of analyzing trial outcomes are shown in Table 3.
2.14.1. Follow-Up Visit Schedule
In the remission induction phase, patients were evaluated at baseline; at weeks 1, 2, 3, and 4; and then at months 2, 4, and 6. In the remission maintenance phase, follow-up visits occurred every 3 months. In the best medical judgment phase, patients had study visits every 6 months until the common closeout date.
2.15 . Outcomes
We designated primary, secondary, and tertiary outcome measures (Table 5).

2.15.1. Primary Outcome Measure
The primary outcome measure was a comparison of the percentage of patients in the two treatment arms who had a BVAS/WG of 0 and successfully completed the prednisone taper by the 6 month visit.
2.15.2. Tolerance Induction
The induction of tolerance to the ANCA antigens was a secondary outcome in RAVE. We adopted a clinical definition of tolerance: (1) complete remission without the use of immunosuppressive therapy, including glucocorticoids; (2) reconstituted peripheral blood B cell count (return to a level of absolute CD19+ cells at least 80% of baseline); (3) absence of ANCA in the serum; and (4) normal response to neoantigens.
2.16. Sample Size and Power
2.16.1. Statistical Assumptions
We made the following assumptions in calculating the sample size. First, we estimated that 70% of patients in the CYC group would be in remission and off prednisone at 6 months. This assumption was based upon data from the Granulomatosis with polyangiitis (GPA) Etanercept Trial [10], a study in which 66% of patients with severe disease receiving CYC achieved steroid-free remissions at 6 months [WGET Research Group, unpublished data]. Second, a non-inferiority margin of 20% on the difference in the complete remission percentage between the investigational therapy arm and the standard therapy arm was accepted (see below). Third, we planned to use a one-sided 0.025 alpha-level Chi-square test. Finally, a 10% dropout rate was anticipated. These assumptions required 100 patients in each arm (i.e., a total of 200 patients) to have approximately 80% power to reject the null hypothesis of inferiority of RTX.
To conclude non-inferiority of RTX, two requirements had to be met. First, the lower bound of the two-sided 95% confidence interval on the difference between the RTX- versus CYC-treated patients achieving the primary outcome had to be greater than the -20% non-inferiority margin. Second, if the remission rate of RTX was lower than that of CYC, the lower bound of the confidence interval in the CYC arm had to be above 40%, to ensure that the control treatment had been reasonably effective.
The protocol further defined two conditions under which superiority could be concluded. First, the lower bound of the confidence intervals on the difference between RTX and CYC success rates had to be >0. Second, the lower bound of the confidence interval of the RTX arm had to be >50%.
These conditions were established in pre-trial discussions with the Rheumatology Working Group of the FDA. A non-inferiority trial design with a lower limit of efficacy of 50% for the RTX arm – assuming a 70% efficacy in the CYC arm and thereby applying a non-inferiority margin of 20% – was considered appropriate based on the following rationale. The only study documenting outcomes of patients with untreated severe GPA, derived from the pre-CYC era, showed that only 50% of patients were alive 6 months after diagnosis [5]. Under the conservative (albeit highly improbable) assumption that all patients alive in that study had achieved a spontaneous complete remission, RTX therapy could be assumed to be effective for severe AAV if the lower bound of the confidence interval for the RTX group in the trial were higher than 50%. The achievement of complete remission in more than 50% of patients with a drug that potentially offered a better toxicity profile than CYC was felt to be clinically important.
2.17. Statistical Analysis
2.17.1. Primary Efficacy Analysis
The primary efficacy analysis was performed when all patients enrolled had completed their 6 month evaluations. An appropriate α adjustment was made to account for the Data and Safety Monitoring Board’s review of the efficacy margin after the first 100 patients had been enrolled and followed for 6 months. We defined in advance superiority in efficacy as an analysis we would perform if the conditions for non-inferiority were met.
2.17.2. Adverse Events Analysis
We focused on the cumulative occurrence of nine major adverse events, selected in consultation with the FDA. These pre-specified adverse events included side effects of treatments in both groups that were anticipated to be important in assessing the relative merits of the two treatment approaches. The events included: (1) Death (all causes); (2) grade 2 or higher leukopenia or thrombocytopenia (white blood cell count < 3,000/mm3 or platelet count < 80,000/mm3); (3) grade 3 or higher infections; (4) hemorrhagic cystitis; (5) malignancy; (6) venous thromboembolic event (deep venous thrombosis or pulmonary embolism); (7) hospitalization resulting either from the disease or from a complication due to study treatment; (8) infusion reactions that occurred within 24 hours of infusion and resulted in the cessation of further infusions; and (9) cerebrovascular accidents. The numbers of these events per patient-year of follow-up in the two treatment groups was compared at 6, 12, and 18 months.
2.18. Monitoring
2.18.1. Data and Safety Monitoring Board
The Data and Safety Monitoring Board (DSMB) for RAVE was the committee formed by the Division of Allergy, Immunology, and Transplantation (DAIT) at NIAID to oversee clinical trials in autoimmunity. This committee is comprised of academic physician-investigators, clinical trialists, and biostatisticians appointed by the trial sponsor, NIAID. The specific identities of the individuals on this committee are not revealed, so as to avoid potential biases.
During the review process, the DSMB was blinded to specific treatment assignments. However, the DSMB could vote to permit unblinding if necessary to ensure patient safety. The DSMB was authorized to recommend stopping the trial at any time if evidence of an extraordinary difference in efficacy emerged or an important disparity in safety occurred. However, the DSMB did not establish hard stopping rules related to either efficacy or futility.
2.18.2. Adverse Events Reporting
Adverse events were graded according to the National Cancer Institute Toxicity Grading Scale [31].
2.19. Mechanistic Studies
The narrow mode of action of RTX provided unique opportunities for mechanistic studies that address the central hypothesis related to tolerance; namely, that tolerance to the self-antigens PR3 or MPO can be restored by anti-CD20 therapy because of the elimination of PR3- or MPO-specific memory B cells.
2.19.1. Sample Collection
Whole blood flow cytometry was performed centrally at the time of each visit to evaluate the development, frequencies, and activation status of B and T cells, natural killer cells, and myeloid and dendritic cells. The flow cytometry panel is shown in Table 6.
Peripheral blood mononuclear cells (PBMC) were prepared at each visit. We plan to perform PBMC-Elispot assays to determine the proliferative responses as well as antibody and cytokine production of B and T lymphocytes upon antigen stimulation.
Serum and plasma samples were also collected at each visit for batch measurements of ANCA, B cell lymphocyte stimulator (BLyS/BAFF), other cytokines, and future ancillary studies. Both high and low molecular weight DNA was harvested from peripheral blood for studies designed to understand genetic predictors of clinical outcomes. Whole-blood RNA was stored for investigations of gene expression profiling and the identification of associations with treatment response, specific drug resistance and toxicities, and disease relapses.
2.19.2. ANCA Assays
ANCA assays were performed using complementary testing methods, including standard immunofluorescence and direct and capture enzyme immunoassays [32-35].
2.19.3. Vaccine Study
In order to assess patients’ immune responses to recall and neoantigens, we designed a vaccine substudy to assess both cellular and humoral immunity by testing responses to both T cell-dependent and T cell-independent antigens. Two T cell-dependent vaccines were used: (1) the Candida and tetanus (booster formulation) skin test as T cell-dependent recall antigens. (2) the Haemophilus influenzae type b vaccine. Unconjugated pneumococcal vaccine was used as T cell-independent vaccine. Serum for measurements of the antibody responses to the vaccines was obtained at baseline, before immunization, and at 4–6 weeks after immunization. Participation in this vaccine study was optional and administered under separate informed consent in patients who had fulfilled the other components of the tolerance definition after month 18.
3. RESULTS
3.1. Enrollment
The initial concept proposal for RAVE was submitted to the ITN in November, 2002. Funding was granted in May, 2003. Protocol development was completed in the fall of 2004. The first patient was enrolled on December 28, 2004. The 197th patient was enrolled at the end of June, 2008. The decision was made to stop enrollment at that time because the observed drop-out rate was lower than anticipated.
3.2. Challenges
The effects of both CYC and RTX posed potential for unblinding. To conceal the diffuse alopecia that is often associated with CYC use from the investigators who assessed clinical outcomes, all patients wore hats to their study visits for 6 months following the discontinuation of CYC or CYC placebo. Laboratory results such as leukopenia or macrocytosis could also have led to unblinding. Thus, each subject was evaluated by two clinicians, a Safety Officer and an Investigator. The Investigator, who performed the BVAS/WG assessment, viewed only redacted reports of laboratory results, with white blood cell count, lymphocyte count, and mean corpuscular volume values blacked out by the site Trial Coordinator. Mild infusion reactions attributable to RTX occur in approximately 10% of patients [Rituximab Investigator’s Brochure] and infusion reactions can also occur among placebo-treated patients. Thus, the occurrence of an infusion reaction did not necessarily unblind the patient’s treatment assignment. To prevent unblinding of the Investigator, the Safety Officer evaluated and treated all patients with possible infusion reactions. Moreover, subjects were asked to report potentially CYC- or AZA-related adverse events such gastrointestinal symptoms only to the Safety Officer.
AZA was used in lieu of CYC in the remission maintenance phase of the trial. Subjects’ ability to metabolize AZA is determined by the presence or absence of alleles for thiopurine methyltransferase (TPMT) [36]. Because the time required to receive the results of TPMT testing would have constituted an unacceptable delay before initiating trial therapy, TPMT alleles were tested at entry but randomization proceeded before the results became available. Patients heterozygous for TPMT continued in the protocol but at a reduced dose of AZA/AZA placebo. Patients who were homozygous negative for the TPMT allele were unblinded at 6 months and treated thereafter according to best medical judgment.
3.3. Summary of Efficacy and Adverse Events Results to Date
The six-month primary outcome data have been published [37]. The two treatment groups were equivalent at baseline with regard to baseline disease activity, organ involvement, and the proportion of patients with new disease at entry. Sixty-three (64%) of patients assigned to RTX achieved the primary outcome as compared with 52 (53%) in the CYC arm, meeting the criterion for non-inferiority (p<0.001). The superiority comparison for RTX versus CYC fell short of statistical significance (p = 0.089), but RTX was more efficacious than CYC for remission induction among the subset of patients with relapsing disease at trial entry: 34/51 (66.7%) patients allocated to RTX achieved this measure, compared to 21/50 (42.0%) randomized to CYC (p=0.01). This finding persisted even after controlling for differences in ANCA type and clinic site (odds ratio 1.40; 95% confidence interval [RCI] 1.03, 1.91; p=0.03). RTX was also as effective as CYC for treating subsets of patients regarded as having the most severe disease; namely, patients with either major renal disease or alveolar hemorrhage. There were no differences between treatment groups in adverse event (AE) rates, serious AE rates, or non-disease related AEs at 6 months [37].
3.4. Ongoing Analyses
Analyses of data pertaining to treatment efficacy and adverse effects beyond six months, extending to 18 months of follow-up and beyond, are ongoing. Mechanistic studies on samples up to 18 months and beyond are also under way.
4. DISCUSSION
4.1. Equipoise
Despite the fact that CYC has been established in the late 1960s as the standard of care for remission induction in AAV, strong equipoise existed for RAVE. The conventional regimen of CYC and high-dose glucocorticoids has well-recognized toxicities and is ultimately unsuccessful in maintaining remission for the majority of patients [10]. Preliminary data on the use of RTX in AAV were promising [22-24], and the early studies of RTX used strict definitions of remission with a validated instrument [28]. Further uncontrolled studies were felt unlikely to augment the knowledge base related to the safety of rituximab, and such studies are not reliable tests of efficacy. A randomized, double-blind trial was the only way to achieve the dual goals of testing RTX rigorously and determining the impact of this therapy on tolerance induction.
4.2. Safety Checks for Challenging the Standard of Care
Because of concern among the Research Group related to treating severe AAV without CYC, we built into the protocol several provisions designed to protect patients in the event that the hypothesized efficacy of RTX proved unfounded.
- Frequent follow-up after randomization. This feature was inherent in the design of RAVE, because patients had to be evaluated clinically for at least four consecutive weeks to receive their treatment infusions. In addition, patients were evaluated for a strictly-defined disease response by the 1 month visit in order to continue on their assigned therapy (See Disease Response, Section 2.11).
- Careful definition of early treatment failures (Table 1). The DSMB was instructed to recommend stopping the trial if an unexpected number of such events occurred among the first patients enrolled.
- Treatment with glucocorticoids. Patients in both arms were treated with pulse methylprednisolone. High doses of glucocorticoids were expected to temper the manifestations of severe AAV. Thus, the glucocorticoid regimen provided some effective therapy during the period when the CYC and RTX are only beginning to exert their effects.
- Availability of crossover. Blinded crossover was available for patients who experienced severe disease flares during the remission induction phase.
- Physician judgment. Trial physicians were allowed to discontinue patients from the trial at any time if they believed that treatment on the basis of best medical judgment was in the patients’ best interest.
- Exclusion of patients with AAV with the most severe involvement of the kidneys or lungs. Patients with serum creatinine concentrations > 4.0 mg/dL or on mechanical ventilation were not eligible for RAVE.
4.3. Open-Label RTX During Remission Maintenance
The decision to treat severe disease flares in the remission maintenance phase with open-label RTX deserves comment. If a patient (randomized to either RTX or CYC) had achieved disease control and completed the first 6 months of the trial, then the therapy to which the patient had been randomized was successful in inducing remission.
Assuming then that the patient had been randomized originally to CYC, the readministration of CYC to induce remission would have subjected the patient to a significantly increased risk of treatment-associated morbidity. The side effects of CYC relate strongly to the dose and duration of therapy [11,16,38]. Given the equipoise of this trial, an attempt to induce remission with RTX in that setting was appropriate. On the other hand, if the patient had been randomized originally to RTX, a second administration of RTX was reasonable because of the indications from clinical studies in other diseases that the side effect profile of RTX might be superior. Moreover, information to date from the use of RTX in non-Hodgkin’s lymphoma and rheumatoid arthritis indicates that the administration of a second course of RTX is as safe as the first course and not associated with loss of efficacy [39-42].
4.4. Impact of the Funding Source
The funding source for the trial had an important impact on the design of RAVE. The overarching aim of the ITN is to develop therapies for immunologically-mediated disorders, inflammatory diseases, and organ transplantation that restore tolerance. This ITN focus had two implications for the design of RAVE.
First, although remission induction and successful glucocorticoid taper by 6 months were designated as the primary outcome, secondary outcome measures related to immune tolerance necessitated follow-up for a period significantly longer than 6 months. The minimum follow-up of 18 months in this trial offered the opportunity to understand the effects of RTX administration in a variety of clinical scenarios, e.g., after a previous course of RTX or following the first induction of remission with a regimen involving CYC.
Second, the desire to understand the impact of RTX on immune tolerance meant that second courses of this medication could not be built routinely into the protocol. Indeed, with regard to AAV, the optimal timing of RTX readministration, if warranted, was not clear at the time the trial was designed and remains unclear at the time of this writing.
Genentech staff had a consultative role in study design and participated in a pre-trial meeting with the FDA related to the potential labeling of RTX for remission induction in AAV. Genentech representatives also approved the trial protocol, participated in regular conference calls of the Study Management Team, and provided safety updates on RTX studies in other immunologic indications such as rheumatoid arthritis. However, Genentech/Biogen IDEC staff had no direct input into the procedures for data collection or the analysis of trial data, and no decision-making rights regarding what data to publish or when to publish it.
4.5. The RAVE Results in Context
The RAVE trial marked a major challenge to CYC as the cornerstone of remission induction therapy in severe AAV. No head-to-head comparison of any agent against CYC for patients severe AAV had ever been attempted before the RAVE trial. The primary outcome results of the trial demonstrated that treatment with RTX and glucocorticoids is not inferior to the standard regimen in patients with new-onset severe AAV. The trial also showed that RTX and glucocorticoids are superior to the standard regimen of CYC and glucocorticoids for remission induction in severe relapsing AAV.
The RAVE results reported to date focus exclusively on remission induction but do not address the question of retreatment. RTX was not re-administered following the return of peripheral blood B-cells (anticipated by 9-12 months). Longer follow-up in the present trial is important to understand the benefits and risks of RTX therapy for AAV. Analyses of patients followed for a minimum of 18 months will explore issues such as the need for re-treatment, particularly in the context of peripheral B-cell reconstitution.
4.6. FDA Discussions
The acceptance of CYC as the standard of care for remission induction in severe AAV for several decades is based on results from large uncontrolled cohort studies and expert opinion. However, no medication (including CYC) has ever been approved by the FDA for the treatment of AAV. This fact posed a hurdle in the design of this non-inferiority trial and is a problem encountered commonly by investigators who intend to design trials that challenge the “standard of care” in any condition when no such standard is recognized in the form of an FDA-approved regimen.
Because of the lack of data from randomized, controlled trials on the effect of CYC in AAV, the FDA required us to demonstrate not only the relevance of proving non-inferiority to CYC, but also superiority to the expected rate of primary outcome achievement in untreated patients. This point was more difficult to demonstrate, because of the paucity of data on the natural history of untreated disease in the modern era. However, case series published in 1958 [5] and 1967 [43] were instrumental in showing that the lower limit of the confidence interval proposed for our primary outcome measure – set at 50% of the patients in the RTX group achieving a BVAS/WG of zero at 6 months (off glucocorticoids) – would far exceed the anticipated remission rate in patients with severe AAV treated with placebo.
Key elements of our discussions with FDA about the selection of the appropriate non-inferiority margin, primary outcome selection, and demonstration of efficacy of a novel therapy in this rare disease have been published in a Draft Non-inferiority Guidance Document for Industry. (http:// www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM202140.pdfhttp://www.fda. gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM202140.pdf).
Important discussion points in the development phase of RAVE were the orphan disease status of AAV and the practical implications of this status for sample size. The FDA Guidance Document indicates that flexibility in the design of non-inferiority trials may be given by the FDA when: 1) the difference between the active control response and the response of untreated patients is large; 2) the primary endpoint does not involve an irreversible outcome; 3) the experimental treatment is associated with fewer serious adverse events than currently available therapies; or, 4) when the experimental medication is in a new pharmacologic class and is shown to be better tolerated than other available treatments. The design of the RAVE trial aligned with all of those elements and thus represents a paradigm for designing a pivotal trial in an orphan disease indication.
CONFLICT OF INTEREST
Drs. Brunetta and Zhang are employees of Genentech.
Appendix


The RAVE Research Group

ACKNOWLEDGEMENT
RAVE was supported by the Immune Tolerance Network (ITN), which is funded in turn by the National Institute of Allergy and Infectious Disease (NIAID). RTX, matching placebo, and partial funding were provided by Genentech, Inc. (South San Francisco, CA) and Biogen IDEC, Inc. (Cambridge ,MA).