
The Open Drug Metabolism Journal
(Discontinued)
ISSN: 1874-0731 ― Volume 5, 2011
Effects of Rifampin on the Disposition of Gepirone ER and Its Metabolites
Y.W Francis Lam1, 2, 4, *, Larry Ereshefsky3, Andreas Port5, Cees J Timmer6, Peter Dogterom7
Abstract
Objective:
To investigate the effects of rifampin on the steady-state pharmacokinetics of gepirone and metabolites after multiple dosing of both drugs.
Methods:
24 subjects completed a randomized crossover study with 2 study phases separated by a washout period of at least 4 weeks. The subjects received multiple dosing of gepirone extended-release (gepirone ER) (20 mg daily for 2 days titrated to 40 mg daily for 5 days) with and without concurrent use of rifampin 600 mg daily. Plasma concentrations of gepirone and two principal metabolites were determined for up to 48 hours after dosing on day 7. Urinary 6β-hydroxycortisol:cortisol ratio was also used to assess the extent of enzyme induction during both study periods.
Results:
Rifampin significantly decreased the area under the plasma concentration-time curves (AUC) of gepirone and 3´-OH-gepirone by 95% and 65%, respectively. The peak concentration (Cmax) values also were reduced by 92% and 58%, respectively. On the other hand, there were minimal changes in AUC and Cmax of 1-PP during concurrent use of rifampin. Gepirone dosing did not change the urinary 6β-hydroxycortisol:cortisol ratio, in contrast to a 4.1-fold increase in the ratio with concurrent use of rifampin.
Conclusions:
Rifampin significantly decreased the systemic exposure of gepirone and 3´-OH-gepirone. The likely mechanism is induction of CYP3A4-mediated first-pass metabolism in the intestine and the liver. Concurrent use of potent CYP3A4 enzyme inducers might lead to significant reduction in pharmacologic effect of gepirone. On the other hand, gepirone does not appear to have CYP3A4 induction or inhibition effects.
Article Information
Identifiers and Pagination:
Year: 2009Volume: 3
First Page: 1
Last Page: 7
Publisher Id: TODMJ-3-1
DOI: 10.2174/1874073100903010001
Article History:
Received Date: 31/3/2008Revision Received Date: 18/11/2008
Acceptance Date: 24/11/2008
Electronic publication date: 16/1/2009
Collection year: 2009
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Department of Pharmacology, University of Texas Health Science Center at San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229-3900, USA; E-mail: lamf@uthscsa.edu
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 31-3-2008 |
Original Manuscript | Effects of Rifampin on the Disposition of Gepirone ER and Its Metabolites | |
INTRODUCTION
Gepirone is an antidepressant drug belonging to the azapirone class of compounds. It has agonistic activities on 5-HT1A receptors and down-regulates the 5-HT2 receptor but is devoid of affinity for benzodiazepine or dopamine receptors [1]. Clinical studies suggest that gepirone is safe and effective in treatment of anxiety [2] and depression [3].
Gepirone undergoes extensive first-pass metabolism, and its oral bioavailability is about 15% [4]. In humans, it is metabolized primarily to 1-(2-pyrimidinyl)-piperazine (1-PP) and 3´-OH-gepirone (Fig. 1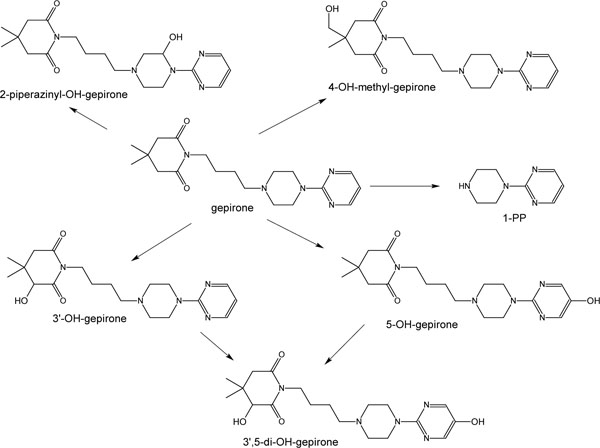 ). Studies in animals indicate that 1-PP may be an anxiolytic, but it is not active in animal models of depression, whereas 3´-OH-gepirone is an anxiolytic [5]. Cytochrome P-450 (CYP) 3A4 plays a role in the formation of 1-PP [6]. In addition, CYP3A4 also accounted for 88% to 92% of the enzyme activity that mediates formation of 3´-OH-gepirone [6]. Therefore, the potential and magnitude of interaction with CYP3A4 modulators needs to be evaluated.
). Studies in animals indicate that 1-PP may be an anxiolytic, but it is not active in animal models of depression, whereas 3´-OH-gepirone is an anxiolytic [5]. Cytochrome P-450 (CYP) 3A4 plays a role in the formation of 1-PP [6]. In addition, CYP3A4 also accounted for 88% to 92% of the enzyme activity that mediates formation of 3´-OH-gepirone [6]. Therefore, the potential and magnitude of interaction with CYP3A4 modulators needs to be evaluated.
 |
Fig. (1) Gepirone metabolic scheme in humans. |
Rifampin is a potent inducer of CYP3A4 and several other CYP isoenzymes within the intestinal wall and in the liver [7] and is involved in numerous drug-drug interactions [8], including those that resulted in significant reduction of plasma concentrations of CYP3A4 substrates [9,10]. The effect of rifampin on the pharmacokinetics of gepirone is not known. The aim of the current study is to investigate the potential and magnitude of an interaction between rifampin and gepirone and its metabolites after multiple dosing of gepirone and rifampin in healthy volunteers. The investigation is a joint effort of study design conception and data analysis in the United States, statistical and analytical input in the Netherlands, and performance of clinical study in Germany.
METHODS
Subjects
Potential volunteers were selected from the records of FOCUS Clinical Drug Development GmbH, Neuss, Germany. 27 nonsmoking healthy volunteers of Caucasian descent (22 men and 5 women; age range, 22 to 45 years; weight range, 53 to 105 kg) participated in the study after giving written informed consent that was approved by the responsible Independent Ethics Committee of the trial center: Ethikkommission der, Düsseldorf, Germany. All subjects were determined to be in good health (on the basis of medical history, physical examination, electrocardiogram, and routine laboratory blood and urine tests) before they were enrolled in the study. None of the subjects received any medication, including any known enzyme inducer or inhibitor, for 1 month prior to first dosing with study drug. They were not allowed to use grapefruit-containing products or St. John's wort for 10 days prior to and throughout the study. All female subjects had a negative pregnancy test and were advised to use a medically acceptable birth control method other than an oral contraceptive.
Study Design
The study involved an open-label, randomized, multiple-oral-dose, two-way crossover design with a washout duration of at least 4 weeks between the 2 study periods. The subjects ingested a 20-mg extended-release formulation of gepirone (20 mg daily for 2 days, followed by 40 mg daily for 5 days), alone (study period A) or in combination with rifampin 600 mg once daily from days 1 to 8 (study period B). For each study period, all subjects were hospitalized in the research unit from 24 hours prior until 2 hours after dosing of study drug on day 1, as well as from the evening of day 6 through completion of pharmacokinetic sampling on day 9. Outpatient visits for study drug dosing and safety evaluation occurred on days 2 to 6.
On day 7 of each study period, each subject received the last 40-mg dose of gepirone ER after a 10-hour overnight fast. Lunch was provided 4 hours after dosing. Timed blood samples of 10 mL each were drawn from the contralateral arm of each subject before and at 0.5, 1, 3, 4, 5, 6, 7, 8, 10, 12, 18, 24, 30, 36, and 48 hours after the day 7 dose of gepirone. All blood samples were processed within 30 minutes to harvest plasma, which was then divided into aliquots and stored at –20ºC until analysis of drug concentrations. To assess attainment of steady-state concentrations in the subjects, predose blood samples of 10 mL each were also obtained on days 5 and 6. These two predose concentration values were compared with the predose concentration value on day 7 and the 24-hour sample on day 8.
To assess CYP3A4 induction using the endogenous 6β-hydroxycortisol to free cortisol ratio (6β-OHC/FC), 24-hour urine samples were collected prior to first dosing on day 1 and after the last dosing on day 7 of each study period. The urine samples were stored at 4ºC during each collection period. The total urine volume was recorded, and a 20 mL aliquot was stored at –20ºC until analysis.
Drug Analysis
Plasma samples were analyzed for concentrations of gepirone, 1-PP, and 3´-OH-gepirone by liquid chromatographic (LC)-electrospray mass spectrometry, with buspirone as the internal standard for the analysis of gepirone and 3´-OH-gepirone, and 2-aminonorbornane as the internal standard for the analysis of 1-PP. Briefly, for analysis of gepirone and 3´-OH-gepirone, 0.5 mL of plasma was mixed with saturated sodiumtetraborate solution and the internal standard. The samples were then extracted twice with a mixture of n-hexane/chloroform (2:1 v/v). The final organic extract was allowed to dry by evaporation under a gentle stream of nitrogen. The dry residue was reconstituted with 100 µL of a 50:50 acetonitrile/0.05 M ammonium acetate solution, and an aliquot was injected into the LC system. For analysis of 1-PP, the procedure was the same, except that a different internal standard was used and the organic extract was back-extracted with 100 µL of 1% formic acid solution. An aliquot of the aqueous solution was then injected into the LC system.
A Luna® C18 (Phenomenex Inc.) column and a gradient system with flow rate of 600 μl/min were used for chromatography. The samples were analyzed by electrospray mass spectrometry. The product ions of the [M+H]+ion were m/z 121.8 for gepirone, m/z 122.0 for 3´-OH-gepirone, m/z 122.0 for buspirone, m/z 122.2 for 1-PP, and m/z 95 for aminonorbornane. The limits of quantification were 0.1 ng/mL for gepirone and 0.2 ng/mL for both metabolites. Low-, medium-, and high-concentration quality-control (QC) samples for all three analytes were prepared and analyzed with each subject's samples. Coefficients of variation of the different QC samples were in the range of 4.5% to 13.2%. No interfering peaks were observed in chromatograms after rifampin administration.
Urine samples were analyzed for determination of cortisol and 6β-hydroxycortisol concentrations by gradient HPLC. Briefly, 1 mL of urine samples were mixed with the internal standard, 6β-hydroxycortisone, before solid-phase extraction. Following evaporation of the organic phase, the residues were reconstituted in 200 µL of water:ethanol (1:1) mixture and 50 µL was injected into the HPLC system. The analytes were separated by a µBondapak® (Waters Corporation) C18 column and detected at 243 nm. The low-, medium-, and high-concentration QC samples, respectively, were 52.2, 209, and 522 ng/mL for cortisol and 40.8, 204, and 815 ng/mL for 6β-hydroxycortisol. The lower limits of detection were 10.4 ng/mL for free cortisol and 15 ng/mL for 6β-hydroxycortisol. Coefficients of variation of low-, medium-, and high-concentration QC samples were in the range of 1.1% to 7.1% for 6β-hydroxycortisol and 1.3% to 17.8% for free cortisol.
Pharmacokinetic Analysis
Pharmacokinetic parameters of gepirone, 3´-OH-gepirone, and 1-PP, including area under the plasma concentration-time curve from 0 to 24 hours (AUC0-24), apparent clearance (Cl/F), and terminal elimination half-life (t1/2), were calculated using standard noncompartmental methods with the SAS program, version 6.12. The peak plasma concentration (Cmax) and time to peak concentration (Tmax) were determined by examining the concentration-versus-time curves for each subject. The terminal segment of the natural log concentration-versus-time curve for all individual subjects was visually identified. The elimination rate constant, β, was determined by simple linear regression of each subject’s data. Elimination half-life was calculated as 0.693/β. The AUC0-24 was estimated with the trapezoidal rule to the measured concentration at 24 hours. Apparent oral clearance was calculated as administered dose/AUC. Percentage chan-ges in pharmacokinetic parameters for gepirone treatment with or without rifampin coadministration were assessed for each subject.
Statistical Analysis
Repeated-measures analysis of variance (ANOVA) was used to assess whether steady state was achieved for gepirone and the two metabolites after multiple dosing of gepirone for 7 days. All pharmacokinetic parameters in both study periods were expressed as mean ± SD in Table 1. The pharmacokinetic parameters, as well as the AUC ratios (for treatment with/without rifampin) of gepirone and the two metabolites were compared by repeated-measures ANOVA. A P value < 0.05 was considered to be statistically significant.
RESULTS
Demographics
Of the 27 subjects who enrolled in the study, 3 discontinued from the study (2 subjects due to occurrence of adverse events and 1 subject due to a protocol violation). For the 24 subjects completing the study, the mean ± standard deviation values were as follows: age, 33.4 ± 5.4 years; weight 76.6 ± 11.5 kg; height, 179 ± 8.1 cm; and body mass index 23.9 ± 2.3 kg/m2.
Pharmacokinetics
For each study period, steady state for gepirone and the two metabolites was achieved in all 24 subjects after 2 days of administration of 40 mg of gepirone ER. The pharmacokinetic parameters of gepirone, 3´-OH-gepirone, and 1-PP after gepirone treatment with and without rifampin coadministration are summarized in Table 1.
Rifampin significantly reduced the plasma concentrations of both gepirone and 3´-OH-gepirone (Fig. 2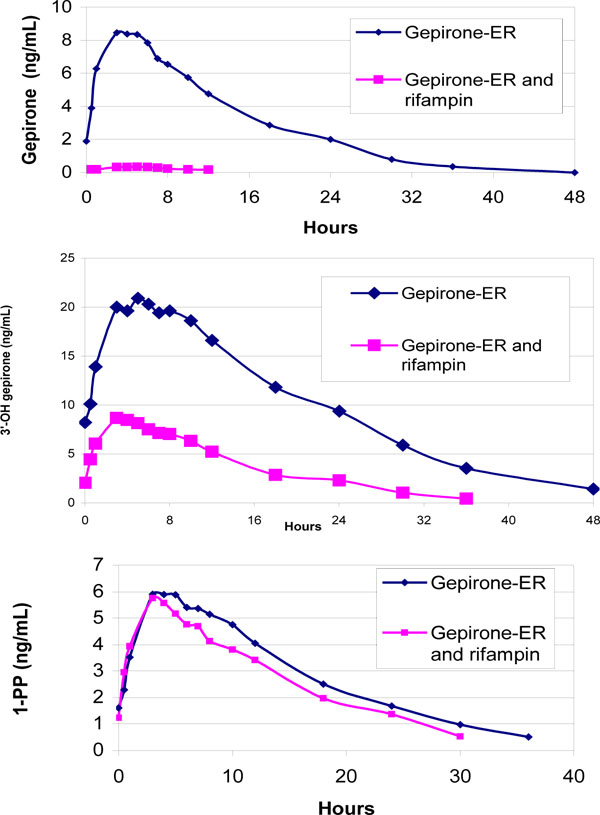 ). The mean Cmax and AUC0-24 of gepirone were reduced by 92.1% ± 9.9% and 94.9% ± 5.2%, respectively. The corresponding reduction in mean Cmax and AUC0-24 for 3´-OH-gepirone was 58.0% ± 11.7% and 64.6% ± 10.5%, respectively. Reductions in Cmax and AUC0-24 were observed for both gepirone and 3´-OH-gepirone in each individual subject. Also, the AUC0-24 for gepirone was decreased by a greater percentage than was the AUC0-24 for 3´-OH-gepirone in all subjects. The AUC0-24 ratio (with/without rifampin) was significantly higher for 3´-OH-gepirone (0.35 ± 0.11) than for gepirone (0.05 ± 0.05).
). The mean Cmax and AUC0-24 of gepirone were reduced by 92.1% ± 9.9% and 94.9% ± 5.2%, respectively. The corresponding reduction in mean Cmax and AUC0-24 for 3´-OH-gepirone was 58.0% ± 11.7% and 64.6% ± 10.5%, respectively. Reductions in Cmax and AUC0-24 were observed for both gepirone and 3´-OH-gepirone in each individual subject. Also, the AUC0-24 for gepirone was decreased by a greater percentage than was the AUC0-24 for 3´-OH-gepirone in all subjects. The AUC0-24 ratio (with/without rifampin) was significantly higher for 3´-OH-gepirone (0.35 ± 0.11) than for gepirone (0.05 ± 0.05).
 |
Fig. (2) Arithmetic mean plasma concentrations of gepirone, 1-PP and 3’-OH-gepirone in 24 healthy subjects after multiple dosing of gepirone (40 mg/day), with or without rifampin therapy (600 mg/day). |
Rifampin also resulted in a 37% reduction in the elimination half-life of 3´-OH-gepirone. For gepirone, the terminal phase was not visually identifiable in 4 subjects during rifampin treatment. During the gepirone-only treatment, 1 sub-ject had an elimination half-life that was more than 15 times longer than that in the other subjects; no clinical and/or analytical reasons were present that could account for the significant discrepancy. Comparison of gepirone elimination half-life values was therefore based only on 23 subjects for the gepirone-only treatment and 18 subjects for gepirone and rifampin co-administration and showed a doubling of the elimination half-life during rifampin treatment. Rifampin shortened the mean Tmax of 3´-OH-gepirone by about 2 hours. There was no significant difference in the Tmax of gepirone with concurrent use of rifampin.
In contrast, concurrent use of rifampin produced only minimal changes in mean Cmax (2.3% ± 23.1% reduction) and AUC (7.1% ± 30.7% reduction) for 1-PP (Fig. 2). There was also much more variability in the direction and magnitude of change in the two pharmacokinetic parameters of 1-PP: 12 subjects had 1.9% to 37.4% increases in Cmax and the other 12 subjects had 0.6% to 61.9% decreases in Cmax. The AUC of 1-PP was reduced in 15 subjects (by 4.2% to 65.5%) and increased in 9 subjects (by 1.9% to 54.7%). In addition, the AUC ratio (with/without rifampin) for 1-PP (0.93 ± 0.31) were significantly higher than that for gepirone and 3´-OH-gepirone. Rifampin had no significant effect on the Tmax or elimination half-life of 1-PP.
CYP3A4 Induction
The baseline total amount of 24-hour urinary 6β-hydroxycortisol prior to first dosing of gepirone was similar in both study periods: 197 ± 117 µg versus 181 ± 75.6 µg, with approximately 8-fold and 4.5-fold interindividual variability, respectively; these findings are consistent with those reported in the literature. With concurrent use of rifampin, the subjects had a more than 4-fold increase in the 24-hour excretion of 6β-hydroxycortisol (Table 2). Similarly, there was more than 4-fold increase in the mean urinary 6β-OHC/FC ratio from baseline. The extent of this increase ranged from 135% to 1100%. On the other hand, dosing of gepirone alone for 7 days only resulted in a 3.1% ± 47.5% mean increase in the 6β-OHC/FC ratio. The extent of change in the ratio is more variable, with a range of 61% decrease to 133% increase. There is also no consistent pattern of incr-ease in urinary excretion of 6β-hydroxycortisol as that demonstrated with rifampin administration in all 24 subjects.

Adverse Effects
Headache, dizziness, and fatigue were the most common adverse effects reported by the subjects. Their occurrence was most frequent during the first couple of days of each study period. Similar frequencies of these adverse effects were observed with and without rifampin treatment. However, four subjects reported moderate (3 cases) to severe (1 case) dizziness on day 1 or day 3 during the gepirone-only phase, as compared to no subjects during coadministration of rifampin.
DISCUSSION
This study describes the effects of rifampin on the steady-state pharmacokinetics of gepirone and its two primary metabolites, 3´-OH-gepirone and 1-PP. The AUC of gepirone and 3´-OH-gepirone were significantly decreased in the presence of rifampin. Although there was interindividual variability in the magnitude of changes (80.6% to 99.6% for gepirone and 41.0% to 84.3% for 3´-OH-gepirone), significant reduction in AUC, especially for gepirone, were observed in all subjects. The extremely low gepirone concentrations in the presence of rifampin (Fig. 2) made it difficult to accurately determine the gepirone half-life in the 18 subjects, which likely accounts for the observed unexpected doubling of the elimination half-life during rifampin treatment.
CYP3A4 plays a significant role in the metabolism of gepirone [6]. Significant amounts of this isoenzyme are present within the intestinal wall [11] and in the liver, which probably represent important sites for first-pass metabolism of gepirone. Rifampin has been demonstrated to be a potent inducer of both intestinal and hepatic CYP3A4 [10, 12]. The significant decreases in AUC and Cmax in comparison to the change in elimination half-life of gepirone suggests that the interaction with rifampin is mostly a result of induction of CYP3A4-mediated first-pass metabolism of gepirone in the intestine and in the liver. Since rifampin is also an inducer of intestinal P-glycoprotein expression [13], this mechanism of interaction remains a possibility even though it is unknown whether gepirone is a substrate for P-glycoprotein.
In the current study, gepirone and rifampin were coadministered for 7 days, and the rifampin treatment was continued for an additional day during the pharmacokinetic sampling period. Although the maximum induction of CYP3A4 activity is reportedly reached within 2 weeks of 600 mg daily of rifampin [14], the AUC and Cmax for gepirone in this study were reduced by more than 97% and 95%, respectively. While the magnitude of the interaction effect could be even greater in some subjects with additional days of rifampin administration, the significance of the interaction has been sufficiently demonstrated with the current study design and result.
In vitro data suggest that CYP3A4 is the principal cytochrome P-450 isoenzyme mediating the formation of 1-PP [6]. Detectable 1-PP formation was demonstrated only for recombinant CYP3A4, and ketoconazole produced a concentration-dependent inhibition of 1-PP formation (to as low as <5% of control values) [6]. Although there was significant variability in the magnitude and direction of changes in AUC and Cmax, our study showed there was no significant change in AUC (7.1% ± 30.7% reduction) and Cmax (2.3% ± 23.1% reduction) for 1-PP with concurrent rifampin use. These reductions were significantly smaller for 1-PP than for gepirone or 3´-OH-gepirone (Table 1). Despite the significant changes in the 6β-OHC/FC ratios after rifampin co-administration, there was no correlation between the urinary 6β-OHC/FC ratios and the 1-PP to gepirone AUC ratio after rifampin co-administration (r2 = .0026).
There are numerous reasons (e.g. inability to assess effect of liver blood flow, protein binding, and extent of liver partitioning) why in vitro metabolic results using human liver microsomes do not necessarily correlate with those from in vivo pharmacokinetic study. The discordance between our in vivo study results after rifampin co-administration and in vitro evidence of CYP3A4-mediated formation of 1-PP [6] suggest other CYP isoenzymes are involved in the formation of 1-PP.
Our findings showed that rifampin reduced the Cmax and AUC of 3´-OH-gepirone, suggesting that rifampin also induced the metabolism of 3´-OH-gepirone, although the enzyme mediating this specific metabolic step is not known. Rifampin can induce several CYP isoenzymes, phase II enzymes, as well as P-glycoprotein [7]. Overall, the induction effect of rifampin on CYP3A4 appears to be more dramatic than for other CYP isoenzymes. An in vitro study [15] has shown greater inducibility for CYP3A4 than for CYP2C9, and clinical studies have suggested similar results. Concurrent use of rifampin 600 mg daily resulted in more than 87% reduction in AUC of CYP3A4 substrates including midazolam, triazolam, simvastatin, and buspirone [9,16-18] compared to 22% to 39% reduction in AUC of CYP2C9 substrates such as fluvastatin, glimepiride, glipizide, and glyburide [19-21] and 50 to 54% for CYP2C8 substrate such as pioglitazone [22,23]. Further studies are needed for confirmation of the specific CYP- or non-CYP-mediated metabolism of 3´-OH-gepirone.
Although renal clearance of cortisol might influence the interpretation of urinary 6β-OHC/FC ratio as a measurement of CYP3A4 induction [24,25], our result showed a dramatic difference between the effects of rifampin versus gepirone on the urinary 6β-OHC/FC ratio within the same subjects. The negligible effect of gepirone on the urinary 6β-OHC/FC ratio suggests that multiple dosing of 40 mg daily of gepirone ER would probably not induce CYP3A4. In addition, even though it is well known that CYP3A5 also contributes to the production of 6β-hydroxycortisol, this specific isoenzyme has not been shown to be inducible [26].
CONCLUSION
Our study suggests that concurrent use of potent CYP3A4 inducers (e.g. rifampin, carbamazepine) might lead to significant reduction in pharmacologic effect of gepirone. However, gepirone evidently did not induce CYP3A4. These metabolic data would have implications in selection of therapy for patients receiving multiple drugs for management of concurrent medical and/or psychiatric conditions.
ACKNOWLEDGEMENTS
We than Jerry Colca (Metabolic Solutions Development) and Patrick Blanner (Pfizer) for providing advice and mitoNEET reagents.