
The Open Drug Metabolism Journal
(Discontinued)
ISSN: 1874-0731 ― Volume 5, 2011
An Experimental Pharmacokinetic Computer Program to Predict Potential Drug-Drug Interactions
Simbarashe Peter Zvada1, 2, Tangai Edwin Chagwedera2, Rosemary Chigwanda2, Collen Mutowembwa Masimirembwa*, 1
Abstract
Polypharmacy as a result of combating co-infections, or combination therapy for better efficacy and reducing the emergency of drug resistance, is on the increase in the African clinical setting in the advent of HIV/AIDS, and tuberculosis (TB) co-infections, and increasing incidences of malaria and other tropical infections. The clinicians and pharmacists are therefore faced with the challenge of prescribing drugs in combinations that are likely to result in severe adverse effects or compromising treatment success. The aim of this study was, therefore, to develop a simple stand alone or network based experimental computational tool to assist doctors and pharmacists in detecting drug combinations likely to result in undesirable metabolism based drug-drug interactions (DDIs) and offer alternate safe prescription options. The mechanism of most drug-drug interactions is through inhibition and induction of drug metabolising enzymes. Models for the prediction of reversible and irreversible inhibitors of the major drug metabolising enzyme system, cytochrome P450, were used in developing the pharmacoinformatic tool. These models enable the prediction of likely in vivo drug-drug interactions from in vitro data. In vivo drug-drug interaction data from the literature was also loaded into the software to validate the system and to give clinical guidance on specific drug-drug interactions. In this first phase of the project, focus was on medications used in the treatment of HIV/AIDS, TB, malaria and other diseases common in Africa. The prototypic tool was based on a Standard Query Language (SQL) database with DELPHI 6.0 as the user interface. Its user friendly pages lead the doctor or pharmacist through drug combination entry functions and gives warning if an interaction is likely. Subsequent actions enable the operator to retrieve more information on the mechanism of interactions, the quantitative measure of the interaction, access to published abstracts on studies, and possible prescription options to minimise DDIs. The software currently has data for 50 drugs used in the design and focuses on the treatment of tropical diseases in addition to classical cases of drug-drug interactions involving other general classes of drugs. The tool can be distributed on Compaq Disk (CD) and be run on any Personal Computer (PC) on windows. We have successfully developed a pharmacokinetic- based tool with a potential to assist clinicians and pharmacists in detecting and rationalizing DDIs. The tool has proved very useful as a teaching tool on DDIs by using the more advanced functions that explore the performance of current drug-drug interactions prediction models. From the available literature, it is clear that more studies need to be done to establish the prevalence and mechanisms of DDIs in the treatment of infectious diseases. We are now adding more data, validating the tool and finally testing the acceptability of this tool among clinicians and pharmacists for routine use.
Article Information
Identifiers and Pagination:
Year: 2009Volume: 3
First Page: 8
Last Page: 16
Publisher Id: TODMJ-3-8
DOI: 10.2174/1874073100903010008
Article History:
Received Date: 2/6/2008Revision Received Date: 29/9/2008
Acceptance Date: 7/11/2008
Electronic publication date: 26/1/2009
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/ which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the DMPK & BAC Department, African Institute of Biomedical Science and Technology, LAPF Centre, P.O. Box 2294, Harare, Zimbabwe; Tel: 263 4 710563; Fax: 263 4 710562; E-mail: collen.masimirembwa@aibst.com, collenmasimirembwa@yahoo.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 2-6-2008 |
Original Manuscript | An Experimental Pharmacokinetic Computer Program to Predict Potential Drug-Drug Interactions | |
INTRODUCTION
In clinical practice, drug prescription error is a common occurrence [1,2] and it results in sub-therapeutic dosing or adverse drug reactions (ADRs). Studies carried out in the United States of America (USA) by the Committee on Quality of Health Care in America (CQHCA) revealed that 44 000 to 98 000 deaths occurring annually are due to medication errors and 7 000 of the total deaths are due to ADRs [3]. It was also estimated that, over 350 000 ADRs occur yearly in United States nursing homes [4]. Again, a retrospective analysis at two London hospitals found that 11% of admitted patients experienced adverse events where 48% were judged to be preventable and 8% led to death [5]. All these studies have given us a hint that the situation might be worse in developing nations where medical expertise and infrastructure are underdeveloped.
The increased demand for pharmacotherapeutic agents in infectious disease co-infections has introduced complexity in the treatment and management of patients in resource limited nations [6]. In the treatment and management of such cases, drug combinations are inevitable as they help in the rapid eradication of causative pathogen, minimize emergence of drug resistant parasites and lead to quick patient recovery. However, each drug provides both therapeutic and toxic effects, thus making physicians worry about safety in the simultaneous use of many drugs. Polypharmacy predisposes patients to adverse drug reactions (ADRs) or adverse drug events (ADEs) emanating from drug-drug interactions (DDIs) [7,8].
Scientifically, a number of clinically significant DDIs occur at the metabolic level. Metabolic drug-drug interaction results in decreased therapeutic effect, generation of toxic metabolites due to enzyme induction or increased substrate plasma concentration above its therapeutic index to toxic levels due to enzyme inhibition. The impact of DDIs is revealed through withdrawals of mibefradil, sirovudine, astemizole and cisapride from the pharmaceutical market [9].
Increased understanding of biochemical pathways of drug metabolism and development of in vitro methods to identify chemical entities that may cause clinically significant DDIs has aided in the screening for safer lead compounds at preclinical stage. Even though this is the case, drug combinations are unavoidable but some drug-drug interactions can be prevented. Recent investigations in the use of drugs have shown that most of DDIs can be prevented through identification of compounds that have in vitro inhibition constant (Ki) of less than 1µM. These chemical entities result in clinically significant DDIs due to enzyme inhibition [10]. Categorically, compounds with Ki value above 10 µM were considered as weak inhibitors. Furthermore, efforts are being made to quantitatively predict the magnitude of in vivo DDIs using in vitro data. However, although very useful, these predictions are being implemented at preclinical levels of drug discovery. Most antiparasitic drugs where discovered and introduced on to the market without knowledge of this data and continue to be used without caution with respect to the potential for drug-drug interactions. Towards addressing this shortcoming, retrospective in vitro screening of antiparasitic drugs for inhibitory and inductory effects on CYP450s were carried out in our laboratory [11-13]. There is, however, still a big gap in clinical evaluation of some of the potential DDI that might arise in the use of various combinations of drugs in the treatment and/or prophylaxis of TB, malaria, HIV/AIDS and other infectious diseases.
Though efforts have been made to assist physicians in handling large patient data and decision making, the available computer based decision support systems like CPOEs offer fragmented data [14,15]. In order to address these deficiencies in health care, the aim of this work was to develop a computer based pharmacoinformatic tool for use in the prediction of drug-drug interaction. Our target groups are clinicians and pharmacists. In this work, CYP450-drug interactions were used as the basis of predicting in vivo DDIs after oral drug administration. Published in vivo data that supports interaction between particular drug combinations was incorporated. Doctors and related users will be assisted in the selection of safer alternative drugs if their first option is not suitable. In addition, the tool will also be used for teaching purposes in preclinical pharmacokinetic concepts towards the prediction of DDIs and appropriate dose estimate utilizing in vitro and in vivo data. Other studies with the same scope exist and they focus on the compilation of a DDI databases [16-18]. The major differences with our study are algorithms used and inclusion of drugs mostly used Africa, e.g. antiparasitic drugs.
In the African setting, internet connectivity is generally slow and expensive making web-based applications unsuitable and unfriendly for some users. However, this can be rectified through our efforts to make the software suitable for both standalone and local area network applications with the possibility of data synchronizations. In order to appreciate most of the functionalities, we recommend hands on use of the prototypic tool both in preclinical and clinical settings.
MATERIALS AND METHODS
Data Collection
Literature sources were assembled including databases, symposia abstracts and original publications on drug-enzyme or drug-drug interaction for both mechanism based and reversible inhibition (Data for the 50 compounds included in this study is available as supplementary material on request). Sources with validated in vitro methods and having the following parameters as well as mechanism of inhibition were considered:
- Inhibition constants(Ki)
- Maximal inactivation rate at saturating inhibitor concentration (kinact)
- Inhibitor concentration at which half maximal inactivation rate is achieved (kI)
- First order rate constant for in vivo gut enzyme degradation (kdegrad.gut)
- First order rate constant for in vivo liver enzyme degradation (kdegrad.hep)
In vivo parameters considered
- Fraction unbound in plasma (fu)
- Fraction of the substrate metabolized in the gut (Fg)
- Fraction of hepatic clearance subject to metabolic inhibition (fm) and fractional contribution of hepatic to total clearance (fh)
- Fraction absorbed from the gut (fa)
- Absorption rate constant (ka)
- Elimination rate constant (kel)
- Time to reach peak plasma concentration (Tmax)
For in vitro studies, types of enzymatic models used are also important, for example, hepatocytes, subcellular fractions and recombinant enzymes have different pros and cons in their predictive value of in vivo DDIs. There was a bias for drugs used in the treatment of tuberculosis, malaria and HIV/Aids in addition to other well-known drug-drug interactions for non-infectious diseases.
Scaling Models
Competitive Inhibition
For competitive inhibition, the following model which has been previously derived for extrapolation of in vitro to in vivo prediction of drug-drug interaction was incorporated [19,20]. The model was applicable to all other CYPs except CYP3A4.
Where CLcontrol/CLinhibitor is the ratio of oral clearance of the drug in the absence and presence of inhibitor respectively, Iin.vivo is the in vivo concentration of inhibitor and AUCinhibitor/AUCcontrol is the ratio of oral clearance of the drug in the presence and absence of inhibitor respectively. Iin,vivo represents various concentrations tested: maximum plasma concentration (Cmax); unbound maximum plasma concentration (fu x Cmax); maximum concentration of the inhibitor at entrance to the liver (Iin,max) and unbound, maximum concentration of the inhibitor at entrance to the liver (fu × Iin,max)
For CYP3A4, there was gut enzyme activity consideration and the following equation was used [21].
Where Fg.inhibitor/Fg.control is the intestinal wall extraction ratio in the presence of the inhibitor in comparison to its absence.
Mechanism Based Inhibition
The following model was considered for all other CYPs except for CYP3A4 [21,22].
(3)
For CYP3A4, there was gut enzyme activity consideration and the following equation was used.
Estimation of Input Parameters
Fraction of Drug Metabolized by Specific Enzyme
An estimate of CYP2C9, CYP2C19 or CYP2D6 contribution to substrate metabolism was predicted using equation 5 [23].
From the above equation, CLpocypPM represents oral clearance in poor metabolizers, and CLpocypEM in extensive metabolizers. This was done where there was no published data. ka, if not given, was estimated from the equation (6) as previously illustrated by Ito et al, 1998 [24]. The result of the calculation must be less than 0.1 min-1 which is the maximum absorption rate constant assuming first order kinetics [25].
Inhibitor Concentration Available to Enzyme In Vivo
Various concentrations can be chosen e.g., unbound, maximum concentration of the inhibitor at entrance to the liver with or without consideration of free fraction. However, the concentration of the inhibitor in the portal vein (Iin.max) was estimated using the following equation as previously described [26].
The value of liver blood flow used was 1470ml/min [24]. Multiplication of equation (7) with fu gives the maximum unbound concentration of the inhibitor at entrance to the liver.
For CYP3A4 inhibitors, effect on the intestinal wall extraction was estimated through the following equation [23].
CLint.g inhibitor/ CLint.g.control represented the first pass intrinsic clearance, and was estimated as follows for competitive inhibition [27].
Mechanism based inhibition was estimated using equation 10 after modification of equation 9.
Iinvivo.g refers to the concentration of inhibitor available to the gut wall absorption site after an oral dose of inhibitor. The dose for estimating the value of the term was estimated through dividing total daily dose by the frequency. The concentration can be estimated as follows using value of 248ml/min [28].
The Pharmacoinformatic Tool Design and Development
a. System Analysis
This is the step where we investigated the applicability of computers in solving the problem. As elaborated in the discussion, medication errors are higher in cases where paper work is being relied upon.
b. System Design
The design of the system was initially done on paper with all the relevant stages and data processing outlined clearly. The mathematical algorithms were detailed in simple English language for easy of programming. At this stage, we evaluated the appropriateness of the available databases and programming languages. A simple and robust tool was proposed.
c. Development
Collected published data was added into the corresponding tables developed in SQL server 2005. Each table had a unique identifier, the key. Depending on the need, tables were linked together through creation of fields that contain same data e.g., the table with drugs’ common profiles had each drug assigned a numerical value which was then linked to its available dosage forms under the table with different fields of dosage forms. This process was done through utilization of SQL commands. The databases architecture created allowed addition, retrieval, storage, back up and audit trail among other features anticipated in all commercial software. The user interface was created using DELPHI 6.0 which was also the programming language. The database and the user interface were linked through a data link utility.
d. Relevance of the Tool
The medical relevance of the tool was tested on selected 5 pharmacists and 5 doctors who have an interest in research. The selection bias was because the types of drugs incorporated into the software were the only ones where published data was available to ease in the evaluation of the predictions.
Assumptions
- The value of Ki was equated to half the IC50 for competitive inhibition if Km equals to substrate concentration
- The fold increase in exposure of the affected drug is related to Ki, Iin.vivo and the fraction of clearance of the affected drug that occurs via metabolism of the inhibited CYP (fm.fh)
- Equal enzyme activity for either in vitro or in vivo
- Fraction absorbed (fa) was assumed to be 1 for rapidly absorbed drugs. In cases where the values were published, factors considered were: dosage form; manufacturer; study population (age groups)
- All the estimation of the fold increase exposure of the affected drug were made assuming a 70kg individual
RESULTS
Development of the software was done successfully. When the user logs into the system, a default page appears. The only difference from Fig. (1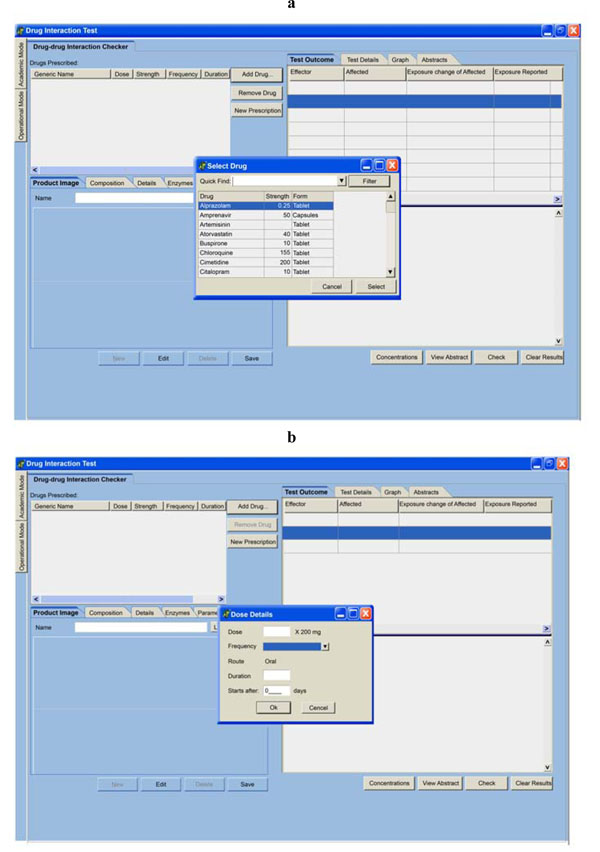 ) is that, the small popped up windows where the physician or pharmacist captures drugs and dosage details will not be displayed but are only prompted by the user.
) is that, the small popped up windows where the physician or pharmacist captures drugs and dosage details will not be displayed but are only prompted by the user.

During routine drug prescription or dispensing by doctors and pharmacist respectively, if they add at least two potentially interacting drugs, a warning is given (Fig. 2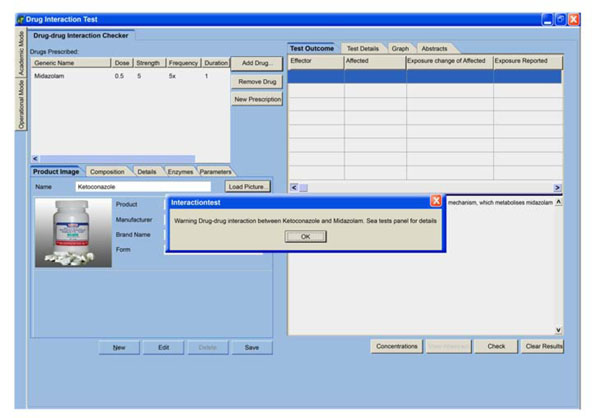 ) followed by a detailed description of interaction (Fig. 3
) followed by a detailed description of interaction (Fig. 3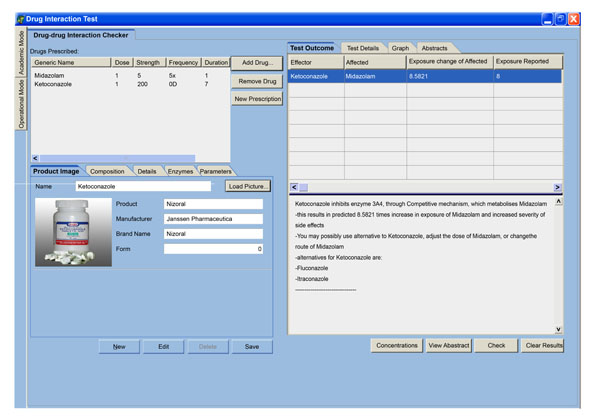 ). The interaction is one-to-many. This means, interaction will be tested on all the drugs already prescribed. The pair in which we have major interactions will be shown in test outcome table. Details of interactions of the added drug with the drug already prescribed can be retrieved through clicking page header, test details, which is next to page header - test outcome. Analysis of different inhibitor concentration was also done to come up with the one that give better prediction on the fold increase in exposure of affected drug when compared with in vivo published data (Fig. 4
). The interaction is one-to-many. This means, interaction will be tested on all the drugs already prescribed. The pair in which we have major interactions will be shown in test outcome table. Details of interactions of the added drug with the drug already prescribed can be retrieved through clicking page header, test details, which is next to page header - test outcome. Analysis of different inhibitor concentration was also done to come up with the one that give better prediction on the fold increase in exposure of affected drug when compared with in vivo published data (Fig. 4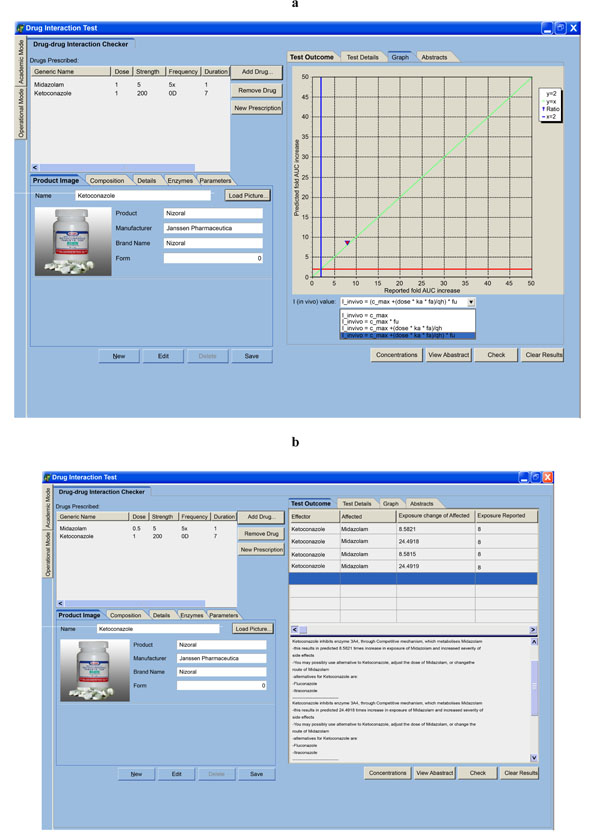 ). Maneuvering of all the pages as highlighted in all figures was done by 5 pharmacists and 5 doctors who expressed support for the enormous potential of the tool in the medical community. At this stage, they recommended that the tool be used as a teaching tool awaiting further validation and capturing of commonly used drug combinations. Should this tool finally find its way into the medical field the best targets recommended are doctors.
). Maneuvering of all the pages as highlighted in all figures was done by 5 pharmacists and 5 doctors who expressed support for the enormous potential of the tool in the medical community. At this stage, they recommended that the tool be used as a teaching tool awaiting further validation and capturing of commonly used drug combinations. Should this tool finally find its way into the medical field the best targets recommended are doctors.
 |
Fig. (2) An illustration of a warning given when two potentially interacting drugs are co-prescribed. |
 |
Fig. (3) A page that details mechanism of interaction, alternatives, fold-increase in exposure of affected drug. |

DISCUSSION
The harnessing of the growing field of information technology into various disciplines has gained much recognition and popularity over the past decades. Large amounts of data generated in the medical fraternity from drug discovery to clinical practice often leaves physicians with a tough task to handle. It is documented that only a small percentage of clinically relevant data that is churned is utilized during routine practice [29]. A number of decision support systems have been developed to match the increase in the amount of data although it calls for a well articulated plan and distinct leadership to achieve success. Most medical informatics tools available are mainly to capture and analyze patient medical data. The few that capture drug-drug interactions, act as electronic version of the hard copies and have been shown to be medically useful in minimizing medication errors [30,31]. Software developed to predict these DDIs exist but are utilized by pharmaceutical companies where they are mainly for drug discovery and development [32]. Most of the software is password protected, and the available trial versions do not give a full understanding the pharmaco-kinetics and pharmacodynamic basis of how the interaction is picked.
In this study, we are mainly focusing on the clinical application rather than early phases of drug discovery. Furthermore, we have addressed the issue of both published and unpublished drug-drug interaction since there is a growing need for evidence-based therapeutic approach (Fig. 3; Fig. 5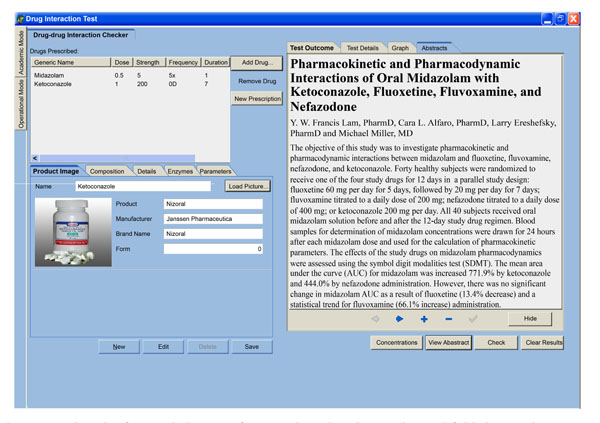 ). These issues make our software different from those already available on the market. Even though a bigger study is yet to be done to test the user friendliness of the interface, savings in time to access DDI data , relevance of warnings and adoption recommendations, tests by a few selected doctors and pharmacists is showing a positive result in that direction. It doesn’t require wide knowledge and expertise in computers, though basic literacy is important. Information retrieval can simply be done through clicking page headers. When the physician or pharmacist logs on in the software, during addition of prescription drugs, dosage details are captured through a small activated window as shown in Fig. (1). Because the user can make mistakes, which is common in the medical field [1,2], the captured drug prescription details are shown on the left corner of the page output allowing for quick and easy editing to make corrections.
). These issues make our software different from those already available on the market. Even though a bigger study is yet to be done to test the user friendliness of the interface, savings in time to access DDI data , relevance of warnings and adoption recommendations, tests by a few selected doctors and pharmacists is showing a positive result in that direction. It doesn’t require wide knowledge and expertise in computers, though basic literacy is important. Information retrieval can simply be done through clicking page headers. When the physician or pharmacist logs on in the software, during addition of prescription drugs, dosage details are captured through a small activated window as shown in Fig. (1). Because the user can make mistakes, which is common in the medical field [1,2], the captured drug prescription details are shown on the left corner of the page output allowing for quick and easy editing to make corrections.
 |
Fig. (5) A page that shows a retrieved referenced abstract of in vivo drug-drug interaction and fold changes in exposure of affected drug. |
If more than one drug is captured and there is a potential for interaction, an interrupting warning message is displayed (Fig. 2). This is important function because it alerts the physician who would otherwise fail to recognize the message if it is shelved or highlighted on other parts of the interface. The message is followed by description of mechanism of interaction, alternative drugs and approaches, and the interacting drugs coupled with the fold increase in the exposure of the affected drug (Fig. 3). It is hoped that physicians as well as pharmacist will benefit from this output both as a teaching tool and in optimal patient health care delivery. Displaying of alternative drugs is based on the magnitude of increase in exposure of affected drug due to the effector. If an alternative to the effector drug causes a higher magnitude, it can not be displayed. This helps in eliminating the trial-an-error approach in choosing alternatives, which is a common practice during routine drug prescription. Detection and minimization of potential DDIs is extremely important in patient care since it reduces chances of adverse drug reactions and events which have been demonstrated to cause ADR, death and result in increased health care costs [5]. Further developments and the use of this tool is hoped to lead to improved interaction between doctors and pharmacists when potential DDIs have been detected in order to give correct drug doses and/or drug combinations to patients.
There is still an on going debate on the best estimate of the correct concentration of the inhibitor available to the drug metabolizing enzyme to use in the models for the prediction of DDIs [27]. Preliminary analysis of different concentrations was done as highlighted (Fig. 4). Recent studies and so far indicated in our prediction tool (Fig. 4), show that better predictive accuracy can be achieved when the unbound concentration of the inhibitor entering the liver from the portal vein was used. We have therefore used this as a default concentration for calculations. The user can also learn how the interaction varies with change in inhibitor concentration as highlighted (Fig. 4) through the use of drop down menu. This also educates users on the possibility of under-prediction or over-prediction when protein binding is taken or not taken into account for the various models. The tool will therefore be responsive to the ongoing advances in research where the role of drug transporters could be important for making better and successful DDI predictions.
In fact, in the medical community pharmacists are not obliged or mandated by any law to alter prescribed drugs but can communicate with the prescriber. Under normal circumstances, patients can either send a relative or a child to purchase medications on their behalf, it becomes difficult for a pharmacist to know the drugs the patient might also be taking in addition to the one being dispensed. With this in mind, pharmacist may not be able to detect potential drug-drug interaction. So the best targets are doctors who came face to face with the patient during disease diagnosis and subsequent drug prescription, and this has been recommended by physicians and doctors. This recommendation supports our hope of leading to improved interaction between doctors and pharmacists when potential DDIs have been detected in order to give correct drug doses and/or drug combinations to patients. In addition, the sparse antiparasitic data that we have uploaded undermines proper validation and routine clinical use.
In conclusion, the tool has the potential to quantitatively predict potential DDIs and could be used by pharmacists and doctors in the minimization of potential DDIs based adverse drug reactions if more in vitro and in vivo data has been added and validated. However more work needs to be done to include DDIs due to induction and irreversible enzyme inhibition or through inhibition of other enzymes not considered in this study.
SUMMARY LIST
What was known before our work
- The qualitative and quantitative nature of drug-drug interaction.
- The contribution of drug prescription error to adverse drug events.
- Need for simple, reliable and easily accessible drug interaction databases.
Contribution of our work
- Detection of potential drug-drug interaction among ARVs, antitubercular and antiparasitic drugs. Most databases do not include most antiparasitic drugs.
- The continued need for simple computer-based decision support systems with ability to predict potential interaction among given combinations of drugs at clinical level.
- Clinical decision support systems with functions on how the decision is reached help in teaching physicians and pharmacist on how to detect potential drug-drug interaction and this improves their knowledge.
- Assistance in picking alternative drugs during drug prescription minimizes the trial-and-error approach, and saves time.
- Electronic systems which enable clinicians to easily access published scientific data improve interaction between researchers and medical personnel.
ACKNOWLEDGEMENTS
Mr Chancemore is gratefully acknowledged for his input in the development of the software; and International, Clinical, Operational, and Health Services Training Award (ICOHRTA) for the fellowship grant for Simbarashe Zvada administered through Biomedical Research Training Institute (BRTI), Harare. We also like to thank doctors and pharmacists who participated in the pilot study. The study was supported by the African Institute of Biomedical Science and Technology (AiBST) Grant 005.