- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Quantitative Proteomic Approaches to Studying Histone Modifications
Barry M Zee, Nicolas L Young, Benjamin A Garcia*
Abstract
Histone post-translational modifications (PTMs) positively and negatively regulate gene expression, and are consequently a vital influence on the genomic profile of all eukaryotic species. The study of histone PTMs using classical methods in molecular biology, such as immunofluorescence and Western blotting, is challenging given the technical issues of the approaches, and chemical diversity and combinatorial patterns of the modifications. In light of these many technical limitations, mass spectrometry (MS) is emerging as the most unbiased and rigorous experimental platform to identify and quantify histone PTMs in a high-throughput manner. This review covers the latest developments in mass spectrometry for the analysis of histone PTMs, with the hope of inspiring the continued integration of proteomic, genomic and epigenetic research.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 5
Issue: Suppl 1
First Page: 106
Last Page: 114
Publisher Id: CCGTM-5-106
DOI: 10.2174/1875397301005010106
Article History:
Received Date: 4/12/2010Revision Received Date: 26/3/2011
Acceptance Date: 25/4/2011
Electronic publication date: 22/8/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at 415 Schultz Laboratory, Department of Molecular Biology, Princeton University, Princeton, NJ 08544, USA; Tel: 609-258-1035; Fax: 609-258-8854; E-mail: bagarcia@princeton.edu
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 4-12-2010 |
Original Manuscript | Quantitative Proteomic Approaches to Studying Histone Modifications | |
THE EMERGING RELEVANCE OF EPIGENETICS FOR GENOMICS
A major goal in genomics is the holistic understanding of a specific cellular process gained through the study of an organism’s genome. An important yet commonly unexplored complement to this analysis is a consideration of the epigenetic influences that impinge on eukaryotic gene expression [1Bernstein BE, Meissner A, Lander ES. The mammalian epigenome Cell 2007; 128: 669-81.]. The first of these pathways is DNA methylation on cytosine nucleotides, which is generally associated with the silencing of imprinted genes. Levels of DNA methylation are maintained by DNA methyltransferases against dilution resulting from DNA replication. Various enzymatic families such as the AlkB family of demethylases, the DME/ROS1 family of glycosylases, and the AID family of cytidine deaminases are also known to remove DNA methylation [2Fritz EL, Papavasiliou FN. Cytidine deaminases: AIDing DNA demethylation? Genes Dev 2010; 24: 2107-14.]. In the specific context of imprinting, several DEMETER-like demethylases localize to the 5’ and 3’ ends of genes and are believed to protect endogenous genes from excessive DNA methylation occurring on nearby transposable elements in A. thaliana [3Penterman J, Zilberman D, Huh JH, Ballinger T, Henikoff S, Fischer RL. DNA demethylation in the Arabidopsis genome Proc Natl Acad Sci USA 2007; 104: 6752-7.]. Another epigenetic pathway involves both small and long non-coding RNAs, where small non-coding interference RNA mediated heterochromatin formation at the centromeres has been documented in S. pombe and D. melanogaster [4Grewal SI. RNAi-dependent formation of heterochromatin and its diverse functions Curr Opin Genet Dev 2010; 20: 134-41.] and that long non-coding RNAs has recently been shown to activate gene expression [5Ørom UA, Derrien T, Beringer M, et al. Long noncoding RNAs with enhancer-like function in human cells Cell 2010; 143: 46-58.].
Finally, epigenetic regulation can proceed via the chromatin associated proteins called histones (the core histone subtypes H2A, H2B, H3, and H4, and the linker histone H1). Although histones were once regarded to be general transcriptional repressors due to their coiling of promoter DNA into the nucleosome, these proteins are currently regarded to have both activation and silencing regulatory influences on gene expression [6Lorch Y, LaPointe JW, Kornberg RD. Nucleosomes inhibit the initiation of transcription but allow chain elongation with the displacement of histones Cell 1987; 49: 203-10., 7Csordas A. On the biological role of histone acetylation Biochem J 1990; 265: 23-38.]. Most histone subtypes exist as multiple sequence variants [8Wang ZF, Sirotkin AM, Buchold GM, Skoultchi AI, Marzluff WF. The mouse histone H1 genes: gene organization and differential regulation J Mol Biol 1997; 271: 124-38., 9Franklin SG, Zweidler A. Non-allelic variants of histones 2a, 2b, and 3 in mammals Nature 1977; 266: 273-5.]. The selective deposition of histone variants may lead to different transcriptional outputs. For instance, while the H3 variant H3.3 is generally linked to gene activation, the H3 variant H3.2 is linked to gene silencing [10Hake SB, Allis CD. Histone H3 variants and their potential role in indexing mammalian genomes: the "H3 barcode hypothesis" Proc Natl Acad Sci USA 2006; 103: 6428-35.]. In addition to occurring as sequence variants, histones can be extensively post-translationally modified with acetylation (ac), methylation (me), phosphorylation (ph), ubiquitination (ub), and other moieties on a residue-specific basis [11Kouzarides T. Chromatin modifications and their function Cell 2007; 128: 693-705.]. Given the broad chemical diversity of modification moieties found on histones, it is perhaps not surprising that a similarly broad diversity of enzymes are responsible for the formation and removal of the modifications. In the case of histone demethylation for instance, at least two distinct enzymatic families exist, namely the flavin- and iron-dependent classes of demethylases [12Shi Y, Whetstine J. Dynamic regulation of histone lysine methylation by demethylases Mol Cell 2007; 25: 1-14.]. How these post-translational modifications (PTMs) are linked to epigenetic regulation is not fully understood. Current models propose that these PTMs act as binding sites for other regulatory proteins that themselves effect transcriptional regulation [13Jenuwein T, Allis CD. Translating the histone code Science 2001; 293: 1074-80.].
Genomics research has uncovered several striking correlations between specific histone PTMs and specific gene regions, mostly using ChIP-Seq. Notable among these correlations is the enrichment of trimethylation of lysine 4 on histone H3 (H3K4me3) over the promoters of active genes and the enrichment of H3K36me3 over the structural coding region of those genes in murine embryonic stem cells [14Mikkelsen TS, Ku M, Jaffe DB, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells Nature 2007; 448: 553-60.]. A recent study applied this correlation and mapped H3K4me3 to the rhesus macaque genome with the aim of identifying novel transcription start sites [15Liu Y, Han D, Han Y, et al. Ab initio identification of transcription start sites in the Rhesus macaque genome by histone modification and RNA-seq Nucleic Acids Res 2011; 39(4): 1408-8.]. A similar approach to identifying novel transcription factor binding sites in murine macrophages has been performed by mapping genomic regions enriched with histone acetylation [16Ramsey SA, Knijnenburg TA, Kennedy KA, et al. Genome-wide histone acetylation data improve prediction of mammalian transcription factor binding sites Bioinformatics 2010; 26: 2071-5.]. Equally interesting discoveries concern the interplay between DNA methylation and histone modifications. For instance, the methylation-specific transcriptional repressor MeCP2 was shown to co-immunoprecipitate with the Sin3 histone deacetylase in X. laevis oocytes. The activity was MeCP2 was further demonstrated to negatively correlate with the overall histone acetylation state [17Jones PL, Veenstra GJ, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription Nat Genet 1998; 19: 187-91.]. Perhaps the strongest lines of evidence linking DNA methylation and histone modification were the isolation of mutations in dim-5 and KYP in screens for DNA methylation defective lines in N. crassa [18Tamaru H, Selker EU. A histone h3 methyltransferase controls DNA methylation in Neurospora crass Nature 2001; 414: 277-83.] and for suppressors of aberrant DNA methylation in A. thaliana respectively [19Jackson JP, Lindroth AM, Cao X, Jacobsen SE. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase Nature 2002; 416: 556-60.]. Subsequent biochemical assays demonstrated that both gene products were in fact histone methyltransferases.
With increasing discoveries of and growing interest towards the functions histone modifications perform for epigenetic regulation, the question arises of how one first discovers and quantifies histone PTMs. Histone modifications were first explored with radiolabeling, as was the case with the biochemical characterizations of the first histone acetyltransferase [20Brownell JE, Zhou J, Ranalli T, et al. Tetrahymena histone acetyltransferase A: a homology to yeast Gcn5p linking histone acetylation to gene activation Cell 1996; 84: 843-51.] and methyltransferase [21Rea S, Eisenhaber F, O'Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases Nature 2000; 406: 593-9.]. Since radiolabeling lacks sufficient specificity for studying PTMs on a residue specific basis, researchers began to rely on the antigenicity of particular PTMs. For instance, indirect immunofluoresence studies determined that K12 acetylation on histone H4 (H4K12ac) is not enriched on any chromosome in D. melanogaster, while H4K16ac is enriched on the male X chromosome over autosomes and the female X chromosomes [22Turner BM, Birley AJ, Lavender J. Histone H4 isoforms acetylated at specific lysine residues define individual chromosomes and chromatin domains in Drosophila polytene nuclei Cell 1992; 69: 375-84.].
Another common antibody-based approach is Western Blotting [23Waterborg JH, Harrington RE. Western blotting of histones from acid-urea-triton- and sodium dodecyl sulfate-polyacrylamide gels Anal Biochem 1987; 162: 430-.]. Western blots helped determine that H2b monoubiquitination is enriched in the more transcriptionally active macronucleus of T. thermophila, while H2A monoubiquitination shows a less dramatic enrichment in the macronucleus over the micronucleus [24Nickel BE, Allis CD, Davie JR. Ubiquitinated histone h2b is preferentially located in transcriptionally active chromatin Biochemistry 1988; 28: 958-63.]. However, with antibody-based approaches such as immunofluoresence, Western Blots, and even ChIP-seq, issues with epitope occlusion and cross-reactivity arise and are difficult to control properly. A specific case of epitope occlusion is the difference in antibody recognition of H3S10 phosphorylation depending on whether the nearby residues H3K9 and H3K14 are unmodified or acetylated [25Cheung P. Generation and characterization of antibodies directed against di-modified histones, and comments on antibody and epitope recognition Methods Enzymol 2004; 376: 221-34.]. In fact, such an influence on PTM binding from the combinatorial influence of nearby residues is predicted to have epigenetic consequences in the form of binary switches. The binding of one protein such as heterochromatin protein 1 (HP1) to methylated H3K9, which can initiate and maintain heterochromatin, may be impeded by phosphorylated H3S10 [26Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond Nature 2003; 425: 475-9.]. A potential case of cross reactivity could be the assignment of biotinylation on histones as novel PTMs [27Kobza K, Camporeale G, Rueckert B, et al. K4, K9, and K18 are biotinylated in human histone H3 FEBS J 2005; 272(16): 4249-59.]. Much of the antibody-based supporting evidence for histone biotinylation has been questioned following a study that determined that streptavidin reactivity could be a possible source of non-specific interaction [28Bailey LM, Ivanov RA, Wallace JC, Polyak SW. Artifactual detection of biotin on histones by streptavidin Anal Biochem 2008; 373: 71-.].
In light of the various shortcomings with radiolabeling and antibody-based approaches, mass spectrometry (MS) is emerging as an experimental platform for identifying and quantifying histone PTMs. Unlike radiolabeling, MS analysis allows for site-specific localization of specific histone PTMs, and unlike antibody-based methods, MS analysis allows for unbiased quantification of histone PTMs. Furthermore, the recent advancements in mass spectrometric analysis are possible to a great extent through parallel advancements in other fields. Thus, this review will not only discuss the latest techniques of mass spectrometry, but also in chromatography, ionization techniques, and in computational software, that have enabled the high-throughput analysis of histone PTMs.
FUNDAMENTALS OF MASS SPECTROMETRY
Before discussing particular MS applications for histone PTM analysis, it is informative to first review the principles of MS analysis. In general, the mass spectrometer measures the behavior of freely moving gas-phase ions in electric and/or magnetic fields. The property measured, such as the time required for an ion to reach the detector, or the frequency of the image current transient induced by an oscillating ion, depends on the mass/charge ratio (m/z) of the ion.
In a mass spectrum, the m/z value of peptide ions are displayed. Given sufficient mass accuracy, from this value alone, one can deduce the composition but not the amino acid sequence of the peptides. For instance, both the sequences PEPTIDE and EDITPEP share the same m/z value (800.368 m/z for the [M+H] singly charged ion). Additionally, peptides with the same sequence containing different sites of modification such as the 9-17 peptide on histone H3 monomethylated (me1) either on K9 or K14 and unmodified (un) on the other lysine residue (H3K9me1K14un or H3K9unK14me1 respectively) share the same m/z value (542.312 m/z for the [M+2H] doubly charged ion). Even different modifications such as trimethylation and acetylation give the same nominal mass shift of 42 Da and thus appear identical in a relatively low mass accuracy instrument such as a linear ion trap. The two modifications actually differ by 0.036 m/z, and this seemingly minute difference can be distinguished with a higher accuracy instrument such as a Fourier transform ion cyclotron resonance mass spectrometer or an Orbitrap mass spectrometer [29Olsen JV, de Godoy LM, Li G, et al. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap Mol Cell Proteomics 2005; 4: 2010-1.].
To determine unambiguously the peptide sequence, one needs to isolate the desired peptide ion and fragment the ion into its smaller, typically N- or C-terminus containing fragments. The fragments are subsequently revealed in the MS/MS spectrum (Fig. 2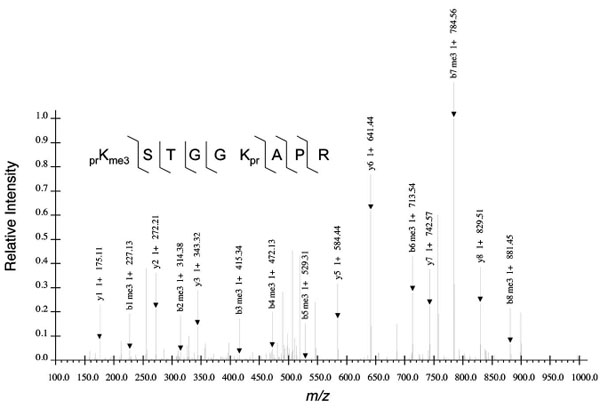 ). The efficiency of fragmentation, signal-to-noise ratio, and other features subsequently influence the confidence of assigning a particular peptide sequence and modification state to a particular MS/MS and to the respective precursor ion in the MS. Such caution is especially warranted given several documented cases of induced migration or scrambling of the methyl group from one histone residue to another during CID fragmentation and leading to PTM misidentification [30Xiong L, Ping L, Yuan B, Wang Y. Methyl group migration during the fragmentation of singly charged ions of trimethyllysine-containing peptides: precaution of using MS/MS of singly charged ions for interrogating peptide methylation J Am Soc Mass Spectrom 2009; 20: 1172-81., 31Zhang J, Chen Y, Zhang Z, Xing G, Wysocka J, Zhao Y. MS/MS/MS reveals false positive identification of histone serine methylation J Proteome Res 2010; 9: 585-94.].
). The efficiency of fragmentation, signal-to-noise ratio, and other features subsequently influence the confidence of assigning a particular peptide sequence and modification state to a particular MS/MS and to the respective precursor ion in the MS. Such caution is especially warranted given several documented cases of induced migration or scrambling of the methyl group from one histone residue to another during CID fragmentation and leading to PTM misidentification [30Xiong L, Ping L, Yuan B, Wang Y. Methyl group migration during the fragmentation of singly charged ions of trimethyllysine-containing peptides: precaution of using MS/MS of singly charged ions for interrogating peptide methylation J Am Soc Mass Spectrom 2009; 20: 1172-81., 31Zhang J, Chen Y, Zhang Z, Xing G, Wysocka J, Zhao Y. MS/MS/MS reveals false positive identification of histone serine methylation J Proteome Res 2010; 9: 585-94.].

BOTTOM UP MASS SPECTROMETRY FOR HISTONE ANALYSIS
There are various different MS experimental designs that can be performed for histone PTM quantification, each requiring different sample preparation steps, chromatography, and MS/MS fragmentation mode (Fig. 1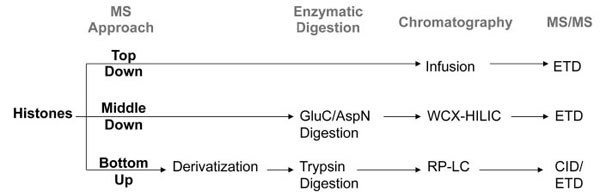 ). Perhaps the most straight-forward approach is bottom up MS analysis. In general, bottom up MS refers to the sequencing and quantification of proteolytic, often tryptic, peptides [32Steen H, Mann M. The abc's (and xyz's) of peptide sequencing Nat Rev Mol Cell Biol 2004; 5: 699-711., 33Bogdanov B, Smith RD. Proteomics by FTICR mass spectrometry: top down and bottom up Mass Spectrom Rev 2005; 24: 168-200.]. Trypsin is widely considered to be the ideal protease for most bottom up applications, including histone analysis, due to its catalytic robustness, relative affordability, and high specificity for lysines and arginines [34Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues Mol Cell Proteomics 2004; 3: 608-14.]. However, the lysine- and arginine-rich histones would be digested by trypsin into very small peptides with poor chromatographic retention. Additionally, the frequent adjacency of lysines and arginines, for example R8 and K9 on histone H3 and K16 and R17 on histone H4, prevents the simultaneous digestion at both residues on the respective proteins. Thus quantification of these histone PTMs would rely on missed-cleavage events that are prone to poor reproducibility. Furthermore, the PTMs themselves often block trypsin digestion, causing missed cleavages for some modified peptides where unmodified or less modified versions of the same peptide digest into normal tryptic peptides. For example, a histone H3 protein unmodified on K9 would produce a different tryptic peptide than another H3 protein trimethylated on K9. While a difference in modification state, such as unmodified and trimethylation, may lead to differences in the ionization efficiencies of both peptides, a difference in the peptide sequence has a greater likelihood to affect the ionization efficiencies even more. This subsequently makes the relative quantification of both modified sites with respect to each other more challenging.
). Perhaps the most straight-forward approach is bottom up MS analysis. In general, bottom up MS refers to the sequencing and quantification of proteolytic, often tryptic, peptides [32Steen H, Mann M. The abc's (and xyz's) of peptide sequencing Nat Rev Mol Cell Biol 2004; 5: 699-711., 33Bogdanov B, Smith RD. Proteomics by FTICR mass spectrometry: top down and bottom up Mass Spectrom Rev 2005; 24: 168-200.]. Trypsin is widely considered to be the ideal protease for most bottom up applications, including histone analysis, due to its catalytic robustness, relative affordability, and high specificity for lysines and arginines [34Olsen JV, Ong SE, Mann M. Trypsin cleaves exclusively C-terminal to arginine and lysine residues Mol Cell Proteomics 2004; 3: 608-14.]. However, the lysine- and arginine-rich histones would be digested by trypsin into very small peptides with poor chromatographic retention. Additionally, the frequent adjacency of lysines and arginines, for example R8 and K9 on histone H3 and K16 and R17 on histone H4, prevents the simultaneous digestion at both residues on the respective proteins. Thus quantification of these histone PTMs would rely on missed-cleavage events that are prone to poor reproducibility. Furthermore, the PTMs themselves often block trypsin digestion, causing missed cleavages for some modified peptides where unmodified or less modified versions of the same peptide digest into normal tryptic peptides. For example, a histone H3 protein unmodified on K9 would produce a different tryptic peptide than another H3 protein trimethylated on K9. While a difference in modification state, such as unmodified and trimethylation, may lead to differences in the ionization efficiencies of both peptides, a difference in the peptide sequence has a greater likelihood to affect the ionization efficiencies even more. This subsequently makes the relative quantification of both modified sites with respect to each other more challenging.

To resolve these histone-specific incompatibilities with trypsin digestion, one must first derivatize the histones. One of the earliest methods was deuteroacetylation, where deuterated acetyl groups are added to all unmodified lysines, and hyperacetylated histone peptides are formed [35Celic I, Masumoto H, Griffith WP, et al. The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation Curr Biol 2006; 16: 1280-9.]. ‘Hyperacetylated’ peptides denote peptides where every lysine is acetylated. Under reversed phase chromatography, all hyperacetylated peptides of the same primary sequence co-elute at effectively the same retention time. While this derivatization seemingly erases the original modification patterns on the histones, in fact the difference in mass between the original and deuterated acetyl groups (Δm = 3.018 Da) allows one to determine the original number of acetyl groups. Deuteroacetylation continues to be an effective approach and was recently used to determine the changes in histone PTM levels resulting from incubating 293T cells with derivatives of pyridine-2,4-dicarboxylic acid, which inhibit the JMJD2 methyltransferase [36Mackeen MM, Kramer HB, Chang KH, et al. Small-molecule-based inhibitio of histone demethylation in cells assessed by quantitative mass spectrometry J Proteome Res 2010; 9: 4082-92.].
Another method of derivatization relies on propionic anhydride to derivatize histones [37Syka JEP, Marto JA, Bai DL, et al. Novel linear quadrupole ion trap/FT mass spectrometer: performance characterization and use in the comparative analysis of histone H3 post-translational modifications J Proteome Res 2004; 3: 621-.]. In contrast to deuteroacetylation, propionylation occurs on both unmodified and monomethylated lysines as well as the N-terminal amine. The addition of a propionyl moiety (Δ = 56.026 Da) not only prevents trypsin digestion at lysines, but also imparts additional hydrophobicity to the histone peptide. The increase in hydrophobicity enhances the chromatographic resolution between different histone peptides, as well as different modified forms of the same peptide. A second derivatization after trypsin digestion adds another propionyl group to the newly created N-terminus.
A useful variation of this technique is to use deuterated d10-propionic anhydride where the hydrogens on the reagent are replaced with deuteriums, similar to the deuterated d3-acetic anhydride described previously [38Plazas-Mayorca MD, Zee BM, Young NL, et al. One-pot shotgun quantitative mass spectrometry characterization of histones J Proteome Res 2009; 8: 5367-74.]. Here, one can use d10-propionic anhydride after trypsin digestion to label the N-terminus of all the histone peptides and results in an additional mass shift of 5.031 Da, beyond the mass of regular propionic anhydride derivatization. One can then mix two different histone samples, where one is treated with d0-propionic anhydride and the other with d10-propionic anhydride, to quantify the relative difference in histone levels in a single MS experiment by using the ratio between the isotopic pairs. Recently, this differential labeling was used to determine changes in histone methylation and acetylation on multiple residues on H3 and H4 resulting from knock-down of the methyltransferase G9a/GLP1, which previously was known only to methylate H3K9 [39Plazas-Mayorca MD, Bloom JS, Zeissler U, et al. Quantitative proteomics reveals direct and indirect alterations in the histone code following methyltransferase knockdown Mol Biosyst 2010; 6: 1719-29.].
IMPORTANCE OF CHROMATOGRAPHY IN BOTTOM UP AND MIDDLE DOWN HISTONE MS EXPERIMENTS
No discussion on proteomics would be complete without describing the importance of chromatographic separations. The opposite of chromatographic separation is direct infusion of the sample into the mass spectrometer, where the entire sample is introduced into the mass spectrometer at once. Although technically simpler to set up, there are a variety of features that make direct infusion particularly ill-suited for bottom up histone analysis. First, the simultaneous presence of all the various modified histone forms results in ionization suppression of relatively low abundant histone peptides by more abundant histone peptides. Second, the lack of chromatographic resolution means that isobaric histone peptides, such as the unmodified 9-17 H3 peptide (H3K9un, 535.304 m/z) and the monomethylated and monoacetylated 9-17 H3 peptide (H3K9me1K14ac1, 535.304 m/z) to directly overlap with each other. Quantification of these two peptides would rely on deconvolution of the fragment ions in the MS/MS for the 535.304 m/z ion, and is generally a complicated computational problem. Despite these shortcomings, direct infusion has traditionally been extensively used for top down histone protein analysis, for reasons as described later. For bottom up and middle down mass spectrometry, though, the use of reversed-phase and hydrophilic-interaction liquid chromatography respectively are essential in the resolution and quantification of highly modified histone peptides (Fig. 3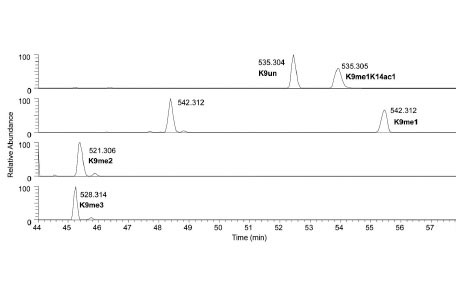 ).
).

MULTIPLEXING IN BOTTOM UP MS FOR COMPARATIVE HISTONE ANALYSIS
To increase the throughput of histone modification analysis, one can use more intricate isotopic labeling schemes for the histone peptides. As already described, d10-propionic anhydride can be used for comparative analysis on different histone samples. Another method for histone labeling is isobaric tag for relative and absolute quantification or iTRAQ. For each sample, a unique isobaric tag is added to all primary amines of the peptide after trypsin digestion, and all the samples are mixed together in equal proportions before mass spectrometry analysis. Each tag is comprised of a reporter and balancer component, and while the net mass of the tag (i.e. reporter1 + balancer1 = reporter2 + balancer2) is constant and equal between differentially tagged samples, the masses of the internal components vary. In the unfragmented full mass spectra, the same histone peptide from the various samples will exactly overlap with each other. The differentiation of the peptides from each sample is determined from the MS/MS spectra, where upon CID fragmentation, the reporter tag is released. It is the relative abundances of the unique reporter tags in a given MS/MS that informs the relative abundances of the histone peptide from each sample.
iTRAQ has so far been used to quantify differences in histone variant protein levels between samples, such as between protein fractions unbound and bound to barrier-to-autointegration factor [40Montes do Oca R, Shoemaker CJ, Gucek M, Cole RN, Wilson KL. Barrier-to-autointegration factor proteome reveals chromatin-regulatory partners PLoS One 2009; 4: e7050.] and between highly tumorigenic cancer lines and their relatively less tumorigenic parental line [41Glen A, Evans CA, Gan CS, et al. Eight-plex iTRAQ analysis of variant metastatic human prostate cancer cells identifies candidate biomarkers of progression: An exploratory study Prostate 2010; 70: 1313-32.]. Despite the promise iTRAQ has for differential PTM analysis, one notable disadvantage of iTRAQ is the reliance of quantification from the MS/MS spectrum. Depending on the efficiency of the precursor ion selection and fragmentation, quantification from the MS/MS spectrum may be more susceptible to noise than quantification from the MS spectrum.
Derivatization with either d10-propionic anhydride or iTRAQ reagents allows for high-throughput histone analysis, and occurs post-sample collection. An alternative mode of histone labeling that occurs pre-sample collection is termed “stable isotope labeling by amino acids in cell culture” or SILAC, which was originally developed for the analysis of the whole proteome rather than only the histones [42Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics Mol Cell Proteomics 2002; 1: 376-86.]. For SILAC, the cells from which histones will be harvested are grown in tissue culture media containing isotopically heavier amino acids such as 13C615N2-lysine, as opposed to the far more abundantly occurring 12C614N2-lysine isotope in nature. If histones are to be harvested from tissue, the organism is fed with a diet where the appropriate amino acid is substituted. By replacing any of the essential amino acids with the heavier isotope and allowing the cells enough time to metabolize the isotope, the histones should fully incorporate the label. Thus, one can mix samples from heavy isotope treated and regular cells together and perform the standard workflow for MS analysis without any further specialized reagents. SILAC has recently been used to understand the PTM profiles on histones H3 and H4 from four human breast cancer lines and a wild type line [43Cuomo A, Moretti S, Minucci S, Bonaldi T. SILAC-based proteomic analysis to dissect the "histone modification signature" of human breast cancer cells Amino Acids 2010. Epub ahead of print].
There are several key limitations to SILAC. For instance, SILAC cannot be performed for human clinical samples for ethical and practical considerations, although as discussed below, newer applications have partly circumvented this dilemma. Both iTRAQ and d10-propionic anhydride can, in principle, be applied to any histone sample regardless of their origin. In addition, the duration of SILAC labeling must be sufficient for near total labeling of the histones. Generally, the slowest turnover for intracellular proteins at steady state is equal to the duration of a single cell cycle, where half of the histone proteins are newly synthesized. Thus, the fraction of remaining metabolically unlabeled histones can be approximated to be (1/2)^n, where n is the number of cell cycles carried out in the experiment. Thus, one must wait until the histones become sufficiently fully labeled before performing any experiments and MS analysis. Additionally, the cost of supplementing isotopically labeled amino acids into tissue culture media may prove more expensive than derivatization approaches.
Despite the shortcomings, one advantage of SILAC for histone analysis is the potentially greater mass shift offered than d10-propionic anhydride, reducing the risk of histone peptide isotope distributions to overlap with each other. 13C615N2-lysine has a mass shift of 8.014 Da, and 13C615N4-arginine has a mass shift of 10.008 Da, and since most bottom up histone peptides contain one lysine and one arginine, one can usually observe a mass shift of over 18 Da. Furthermore, despite the problem with residual unlabeled proteins due to cell turnover, the efficiency of metabolic labeling is generally higher and more easily reproducible than the efficiency of acetic or propionic anhydride derivatization and thus allows the analysis of lower quantity samples [42Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics Mol Cell Proteomics 2002; 1: 376-86.]. Finally, with regard to the incompatibility between SILAC and clinical samples, super-SILAC has been developed as a relatively cost-effective method to circumvent this limitation [44Geiger T, Cox J, Ostasiewicz P, Wisniewski JR, Mann M. Super-SILAC mix for quantitative proteomics of human tumor tissue Nat Methods 2010; 7: 383-5.]. A complex proteomic mixture derived from multiple cell lines cultured in heavy-isotope media is used as a type of standard that can be subsequently spiked into clinical samples [45Geiger T, Wisniewski JR, Cox J, et al. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics Nat Protoc 2011; 6: 147-57.]. The use of multiple lines instead of one increases the likelihood for full coverage of the potentially complex sample proteome, allowing for direct SILAC comparison. In this respect, super-SILAC can be regarded as a proteomic analogue of universal reference RNA commonly used for microarray analysis [46Novoradovskaya N, Whitfield ML, Basehore LS, et al. Universal reference RNA as a standard for microarray experiments BMC Genomics 2004; 5: 20.].
MS APPROACHES TO ANALYZING HISTONE PHOSPHORYLATION AND OTHER PTMs
One of the features that complicate histone PTM analysis is the sheer diversity of modifications found on histones. Traditionally, the diversity of histone PTMs that could be analyzed was limited to the diversity of primary antibodies available. Apart from this technical shortcoming, a larger and more fundamental problem with Western Blots and immunofluorescence is that such antibody-based approaches require a priori knowledge of what modifications to look for. Truly discovery-based screens for novel histone modifications or modification patterns simply could not be performed. Mass spectrometry currently holds the most promise for such endeavors.
Bottom up MS analysis of histone acetylation and methylation is relatively a straight-forward technical problem, with acetylated and methylated peptides usually amenable to standard CID fragmentation. Histone phosphorylation poses a unique challenge for MS analysis, due partly to the general need for enrichment using some variation of metal affinity resin but mostly due to the relatively labile nature of the phosphate bond relative to the methyl or acetyl bond during CID fragmentation. The increased likelihood for loss of the phosphate group from the peptide, and thus the inability to localize the phosphate to a specific residue in the MS/MS, means a different mode of fragmentation is required, namely electron transfer dissociation (ETD). Relative to CID, ETD fragmentation is less biased towards the nature of the amino acid side chains, and by extension the post-translational modification attached to the side chain, and provides a more evenly distributed fragmentation pattern along the peptide backbone [47Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry Proc Natl Acad Sci USA 2004; 101: 9528-33.]. Thermodynamically labile modifications generally remain intact with strong preference for cleavage along the peptide backbone. ETD has proven quite successful in studying histone phosphorylation, with various novel sites identified in H1 variants from asynchronous HeLa cells [48Garcia BA, Busby SA, Barber CM, Shabanowitz J, Allis CD, Hunt DF. Characterization of phosphorylation sites on histone H1 isoforms by tandem mass spectrometry J Proteome Res 2004; 3: 1219-27.] and from mouse and rat testes [49Rose KL, Li A, Zalenskaya I, et al. C-terminal phosphorylation of murine testis-specific histone H1t in elongating spermatids J Proteome Res 2008; 7: 4070-8.]. Given the utility of ETD sequencing of phosphorylated peptides, one may wonder why not all MS experiments operate in ETD mode. Aside from limited availability of ETD in select mass spectrometers, the more crucial limitation is the requirement for higher peptide charge states for effective ETD fragmentation. The combination of chemical derivatization and trypsin digestion usually results in +2 charged histone peptides, which is ideal for CID fragmentation but barely amenable to ETD fragmentation.
In addition to studying histone phosphorylation, mass spectrometry can also be used to study more unique PTMs. The modification of serine and threonine histone residues by β-N-acetylglucosamine was verified using a clever series of chemical derivatization steps to produce a peptide amenable for CID sequencing [50Sakabe K, Wang Z, Hart GW. β-N-acetylglucosamine (O-GlcNAc) is part of the histone code Proc Natl Acad Sci USA 2010; 107: 19915-20.]. A recent survey of protein ubiquitination in a transgenic Arabidopsis line expressing His-tagged ubiquitin monomers also used CID sequencing to identify several novel sites on histone H1.2 [51Saracco SA, Hansson M, Scalf M, Walker JM, Smith LM, RD V. Tandem affinity purification and mass spectrometric analysis of ubiquitylated proteins in Arabidopsis Plant J 2009; 59: 344-58.]. Collectively, these examples illustrate the versatility of mass spectrometry to identify a broad range of histone PTMs.
MIDDLE DOWN MASS SPECTROMETRY FOR COMBINATORIAL PTM ANALYSIS
Thus far, MS analysis on histone PTMs may appear to be restricted to relatively small tryptic peptides that contain 1-4 potential sites of modification. Yet histones are notable for being highly modified proteins that may potentially contain 7 or more modified sites. To understand the connectivity between modifications on different histone residues, one must analyze larger peptides that encompass more modified sites. Bottom up analysis is unable to link, for instance, H3K9me3 to H3K27me3 because both sites occur on separate tryptic peptides. To isolate these larger peptides, alternative proteases to trypsin must be used. GluC or V8 protease digest histone H3 into the 1-50 peptide and encompasses most of the known modified residues on the H3 N-terminal tail except K56 and K79, which have been documented to be acetylated [52Ozdemir A, Spicuglia S, Lasonder E, et al. Characterization of lysine 56 of histone H3 as an acetylation site in Saccharomyces cerevisiae J Biol Chem 2005; 280: 25949-52.] and methylated [53Ng HH, Feng Q, Wang H, et al. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association Genes Dev 2002; 16: 1518-27.] respectively. Middle down MS analysis of the 1-50 peptide can thus link H3K9me3 with H3K27me3, which could not otherwise be accomplished with bottom up analysis (Fig. 4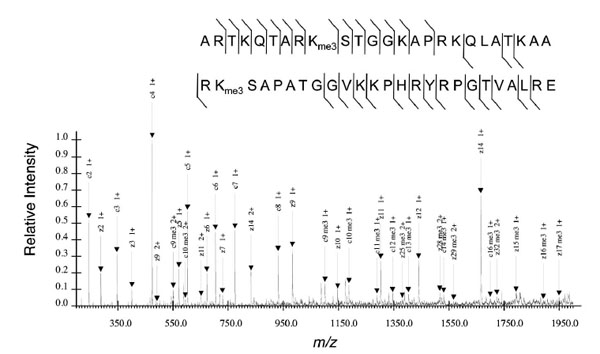 ). AspN digests histone H4 into the 1-24 peptide, and encompasses all the known modified residues on the H4 tail.
). AspN digests histone H4 into the 1-24 peptide, and encompasses all the known modified residues on the H4 tail.

The longer histone peptides, with more lysines and arginines, logically possess higher charge states and are thus more amenable to ETD sequencing than CID sequencing. More crucially though, is that the longer histone peptides require a different mode of chromatography for sufficient resolution of the modified forms. For much the same reasons that propionylated tryptic histone peptides are conducive to reversed phase separation, GluC- or AspN-digested histone peptides are not conducive for reversed phase. In particular, the 1-50 H3 peptide produced from GluC digestion and 1-24 H4 peptide produced from AspN digestion are both highly charged and hydrophilic peptides that would likely elute in the void volume of a reversed phase gradient, as peptides bind and elute based on hydrophobocity. Hydrophilic interaction liquid chromatography (HILIC), essentially the opposite of reversed phase chromatography, is ideal for dealing with these larger and more hydrophilic histone peptides and was initially applied for middle down histone analysis as an offline prefractionation means to resolve different modified forms [54Garcia BA, Pesavento JJ, Mizzen CA, Kelleher NL. Pervasive combinatorial modification of histone H3 in human cells Nat Methods 2007; 4: 487-9.]. A newer variation of HILIC supplements resolution by hydrophilicity with resolution by charge using cation exchange chromatography and allows for direct interfacing with the mass spectrometer [55Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, BA G. High throughput characterization of combinatorial histone codes Mol Cell Proteomics 2009; 8: 2266-84.].
This gain of information on the connectivity between different modified residues on the same protein in middle down mass spectrometry comes at the cost of reduced sensitivity compared to bottom up mass spectrometry. There are at least two reasons for the reduced sensitivity, and both concern an effective dilution of signal. First, a longer histone peptide can exist in a more disperse range of charge states. Bottom up histone peptides mostly exist in charge states of z=1-3 and usually just one or at most two of those is dominant. For larger, more highly charged peptides, such as the 1-50 peptide on histone H3, an broad envelope of charge states is generally observed and four to six charge states can be major contributors to signal [55Young NL, DiMaggio PA, Plazas-Mayorca MD, Baliban RC, Floudas CA, BA G. High throughput characterization of combinatorial histone codes Mol Cell Proteomics 2009; 8: 2266-84.]. Consequently, the overall peptide signal is distributed over more charge states. Second, any gain of information about how PTMs exist in combination comes at the cost of diluting the signal for any one independently considered site. The number of modified versions of a peptide increases as the number of variable sites of modification are considered in combination. For example, there are more than ten of thousand combinations of modifications possible on the intact 1-50 peptide of histone H3. The bottom up mass spectrometry approach collapses these down to tens of modified peptides (including both different sequences and modification states), thus concentrating signal in fewer analytes, but sacrificing molecular connectivity of the PTMs. Just considering the K4 monomethyl species there are over a thousand middle down peptides all of which contain H3 monomethylated at K4 (e.g. H3K4me1K36me1, H3K4me1K9me2K27me2, etc.) but there is only one in the bottom up analysis: H3K4me1. This amount of this one peptide present is equal to the sum of the thousand or more middle down peptides. In reality this effect is much lower than theoretically expected due to high biological specificity of how PTMs exist in combination.
TOP DOWN MASS SPECTROMETRY FOR FULL COMBINATORIAL PTM ANALYSIS
As mentioned, middle down MS analysis achieves nearly full combinatorial analysis on histone H3. One would imagine that the simplest solution to analyze all the modified sites on histone H3 would be to sequence the intact non-digested protein, and indeed, such an approach has already been performed and is termed top down MS analysis. Although not as widely implemented as bottom up or middle down mass spectrometry, top down mass spectrometry has successfully been used recently to discover H2BK37me2 as a novel modified residue in S. cerevisiae and various phosphorylation sites on histone H1 variants, such as H1.4S187ph, enriched on promoters for rDNA and glucocorticoid response elements in HeLa cells [56Gardner KE, Zhou L, Parra MA, Chen X, Strahl BD. Identification of lysine 37 of histone H2B as a novel site of methylation PloS One 2011; 6: e16244., 57Zheng Y, John S, Pesavento JJ, et al. Histone H1 phosphorylation is associated with transcription by RNA polymerases I and II J Cell Biol 2010; 189: 407-15.]. However, the same reasons why middle down MS analysis are less sensitive compared to bottom up analysis are exacerbated for top down analysis, where charge state and combinatorial PTM-dilution effects are compounded when considering the full length protein. In addition, the chromatographic behavior of full-length histone proteins is more difficult to predict than smaller peptides and for this reason direct infusion has typically been used for top down analysis [58Han J, Borchers CH. Top-down analysis of recombinant histone H3 and its methylated analogs by ESI/FT-ICR mass spectrometry Proteomics 2010; 10: 3621-0.]. This approach inherently yields poor resolution of modified forms. This means that the promise of true site specificity and molecular connectivity is often lost when multiple forms that share characteristic ions are present as such mixed spectra usually can not be deconvolved.
Much recent progress in chromatography has been achieved to implement this important step for top down MS analysis. Reversed phase chromatography using ultra-high pressure liquid chromatography has identified very abundant acetylated and methylated forms for all 4 core histone subtypes extracted from immortalized human embryonic fibroblasts, yet lacked sufficient resolution to study modifications on a site-specific basis [59Contrepois K, Ezan E, Mann C, Fenaille F. Ultra-high performance liquid chromatography-mass spectrometry for the fast profiling of histone post-translational modifications J Proteome Res 2010; 9: 5501-9.]. A more extensive survey of intact histone modified forms from human fibroblasts was achieved by resolving proteins first with weak cation exchange-HILIC and second with reversed phase chromatography for two-dimensional HPLC separation [60Tian Z, Zhao R, Toli? N, et al. Two-dimensional liquid chromatography system for online top-down mass spectrometry Proteomics 2010; 10: 3610-20.]. Despite these improvements, more progress in top down histone chromatography is needed for top down histone PTM analysis to accurately and fully characterize the multitude of modified forms that might be observed with sensitivities approaching bottom up or middle down histone studies. Yet, such approaches are appealing as such data would ultimately reflect the complete, fully characterized true identity of the histone proteins, as they exist in nature.
CONCLUSION
Despite important technical limitations particularly with chromatography that remain for MS analysis, mass spectrometry is emerging as a vibrant and versatile experimental platform that can perform comparative PTM analysis between multiple histone samples and even analyze combinatorial PTM patterns. The quantitative rigor and high-throughput characterization of modifications enabled by MS should serve as a valuable complement for genomic studies into the epigenetic role chromatin plays in gene expression.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGEMENTS
B.A.G. acknowledges supported from a National Science Foundation (NSF) Early Faculty CAREER award, an NJCCR SEED grant, an NSF grant (CBET-0941143), and a grant supported by award number DP2OD007447 from the Office Of The Director, National Institutes of Health. B.M.Z is supported through an NSF graduate research fellowship. N.L.Y. is funded by an NIH F32 NRSA postdoctoral fellowship.
REFERENCES
Endorsements
Browse Contents
Table of Contents
- THE EMERGING RELEVANCE OF EPIGENETICS FOR GENOMICS
- FUNDAMENTALS OF MASS SPECTROMETRY
- BOTTOM UP MASS SPECTROMETRY FOR HISTONE ANALYSIS
- IMPORTANCE OF CHROMATOGRAPHY IN BOTTOM UP AND MIDDLE DOWN HISTONE MS EXPERIMENTS
- MULTIPLEXING IN BOTTOM UP MS FOR COMPARATIVE HISTONE ANALYSIS
- MS APPROACHES TO ANALYZING HISTONE PHOSPHORYLATION AND OTHER PTMs
- MIDDLE DOWN MASS SPECTROMETRY FOR COMBINATORIAL PTM ANALYSIS
- TOP DOWN MASS SPECTROMETRY FOR FULL COMBINATORIAL PTM ANALYSIS
- CONCLUSION
- CONFLICT OF INTEREST