- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Open Chemistry Journal
(Discontinued)
ISSN: 1874-8422 ― Volume 8, 2021
Design and Synthesis of a Multi Cu(II)-porphyrin Array
Virginia Gómez-Vidalesa, Andres Borja-Mirandaa, Sandra Cortez-Mayaa, Oscar Amelines-Sarriab, Margarita Riverab, Marcos Martínez-Garcíaa, *
Abstract
A new class of porphyrin array was synthesized. The Cu(II) porphyrin-PAMAM-Cu(II) porphyrin was obtained in good yields. The EPR studies showed that all porphyrins obtained have signals of a copper complex with planar geometry, and no variation in the tetrahedral distortion of the porphyrin ring was observed. The UV-vis and emission studies showed that the intensity of the signal increased due to the presence of 5 porphyrin moieties in the structure. In order to further characterize this molecule, thin films were prepared by using the thermal evaporation method. The morphology of the film showed a homogeneous structure while conductivity measurements exhibit a good conductive response.
Article Information

Identifiers and Pagination:
Year: 2016Volume: 3
First Page: 25
Last Page: 34
Publisher Id: CHEM-3-25
DOI: 10.2174/1874842201603010025
Article History:
Received Date: 1/10/2015Revision Received Date: 27/1/2016
Acceptance Date: 30/1/2016
Electronic publication date: 25/05/2016
Collection year: 2016
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution-Non-Commercial 4.0 International Public License (CC BY-NC 4.0) (https://creativecommons.org/licenses/by-nc/4.0/legalcode), which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Instituto de Química, Universidad Nacional Autónoma de México, Ciudad Universitaria, Circuito Exterior, Coyoacán, C.P. 04510, México city, México; E-mail: margar@unam.mx
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 1-10-2015 |
Original Manuscript | Design and Synthesis of a Multi Cu(II)-porphyrin Array | |
1. INTRODUCTION
The design and synthesis of arrays of oligomeric porphyrins or metalloporphyrins having well-defined shapes and dimensions is currently a topic of great interest to improve the photophysical properties, absorption cross-section and directed energy-transfer reactions [1Chen, C.T. In Comprehensive Supramolecular Chemistry: Coordination Polymers; Pergamon Press: New York, 1996, vol. 5, no. 4, p. 91.-3Ward, M.D. Photo-induced electron and energy transfer in non-covalently bonded supramolecular assemblies. Chem. Soc. Rev., 1997, 26, 365-375.
[http://dx.doi.org/10.1039/cs9972600365] ]. Metalloporphyrin or freeporphyrin arrays with different structures such as dendrimers of arylethynyl chromophores [4Xu, Z.; Moore, J.S. Design and synthesis of a convergent and directional molecular antenna. Acta Polym., 1994, 45, 83-87.
[http://dx.doi.org/10.1002/actp.1994.010450204] ], self-assembling porphyrinic pigments [5Drain, C.M.; Lehn, J.M. Self-assembly of square multiporphyrin arrays by metal ion coordination. J. Chem. Soc. Chem. Commun., 1994, 24, 2313-2315.
[http://dx.doi.org/10.1039/c39940002313] -11Denti, G.; Serroni, S.; Campagna, S.; Juris, A.; Balzani, V. Small-upward approach to nanostructures-dendritic polynuclear metal-complexes for light-harvesting. Mol. Cryst. Liq. Cryst. (Phila. Pa.), 1993, 234, 79-88.
[http://dx.doi.org/10.1080/10587259308042900] ] or metal coordination complexes [12Bothner-By, A.A.; Dadok, J.; Johnson, T.E.; Lindsey, J.S. Molecular dynamics of covalently-linked multi-porphyrin arrays. J. Phys. Chem., 1996, 100, 17551-17557.
[http://dx.doi.org/10.1021/jp961408e] , 13Wagner, R.W.; Lindsey, J.S. A molecular photonic wire. J. Am. Chem. Soc., 1994, 116, 9759-9760.
[http://dx.doi.org/10.1021/ja00100a055] ] containing five, nine or more porphyrins [14Anderson, S.; Anderson, H.L.; Bashall, A.; McPartlin, M.; Sanders, J.K. Assembly and crystal structure of a photoactive array of five porphyrins. Angew. Chem. Int. Ed. Engl., 1995, 34, 1096-1099.
[http://dx.doi.org/10.1002/anie.199510961] -22Solladié, N.; Sooambar, C.; Herschbach, H.; Strub, J.M.; Leize, E.; Van Dorsselaer, A.; Talarico, A.M.; Ventura, B.; Flamigni, L. A photoactive nona-porphyrin with nucleosidic linkers. New J. Chem., 2005, 29, 1504-1507.
[http://dx.doi.org/10.1039/b509913g] ], have been used as an energy funnel, molecular photonic wires [13Wagner, R.W.; Lindsey, J.S. A molecular photonic wire. J. Am. Chem. Soc., 1994, 116, 9759-9760.
[http://dx.doi.org/10.1021/ja00100a055] ] or optoelectronic gates [23Wagner, R.W.; Lindsey, J.S.; Seth, J.; Palaniappan, V.; Bocian, D.F. Molecular optoelectronic gates. J. Am. Chem. Soc., 1996, 118, 3996-3997.
[http://dx.doi.org/10.1021/ja9602657] ]. Recent development in synthesis strategies allows designing a variety of materials, mainly due to the possibility of changing the structure, porphyrin properties, number of porphyrins, interconnections and energy transfer. This diversity is of great importance in the obtainment of light-harvesting devices.
Here, we report a new method to obtain a flexible star PAMAM porphyrin pentamer in order to generate receptors able of binding, recognizing and transferring energy.
2. RESULTS AND DISCUSSION
The porphyrin core 1 and 4 were obtained from para-hydroxybenzaldehyde, benzaldehyde and pyrrole by the conventional method [24James, D.A.; Arnold, D.P.; Parsons, P.G. Potency and selective toxicity of tetra(hydroxyphenyl)- and tetrakis(dihydroxyphenyl)porphyrins in human melanoma cells, with and without exposure to red light. Photochem. Photobiol., 1994, 59(4), 441-447.
[http://dx.doi.org/10.1111/j.1751-1097.1994.tb05062.x] [PMID: 8022886] , 25Vaijanthimala, G.; Krishnan, V.; Mandal, S.K. Cyclic porphyrin dimers as hosts for coordinating ligands. J. Chem. Sci., 2008, 120, 115-129.
[http://dx.doi.org/10.1007/s12039-008-0014-3] ]. The tetrahydroxyporphyrin was used as free based material. Metallation was performed by adding a suspension of copper (II) acetate in methanol to a solution of the free base porphyrin. The tetraester 2 was obtained from methyl bromoacetate with the phenol of the porphyrin 1 in acetone and K2CO3 as catalyst. Finally the amide 3 was prepared with ethylenediamine (Scheme 1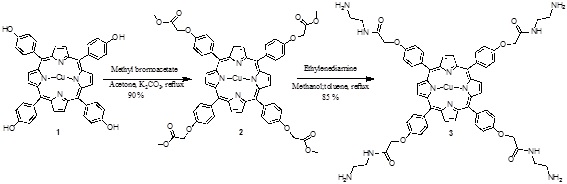 ).
).
The porphyrin 4 was prepared according to reported methodology [25Vaijanthimala, G.; Krishnan, V.; Mandal, S.K. Cyclic porphyrin dimers as hosts for coordinating ligands. J. Chem. Sci., 2008, 120, 115-129.
[http://dx.doi.org/10.1007/s12039-008-0014-3] ] and the copper (II) porphyrin 5 was obtained using Cu(OAc)2, and this was modified with methyl bromoacetate in acetone and K2CO3 as catalyst, obtaining the monoester porphyrin 6 (Scheme 2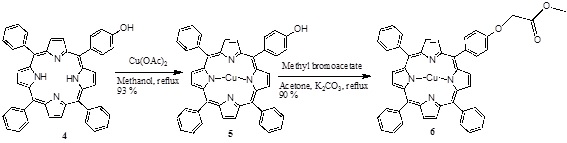 ).
).
The 1H-NMR spectrum of compound 6 exhibits: one singlet at δH 3.74 due the protons of O-CH3 groups, at δH 4.22 due to the CH2-O, two signals at δH 7.27, and δH 7.73 due to the AB system at the para substituted benzene group, and the signals at δH 7.31, 8.21 and 8.83 due to the aromatic and pyrrole protons.
 |
Scheme (1) Synthesis of the porphyrin-PAMAM dendrimer. |
 |
Scheme (2) Synthesis of the mono ester porphyrin. |
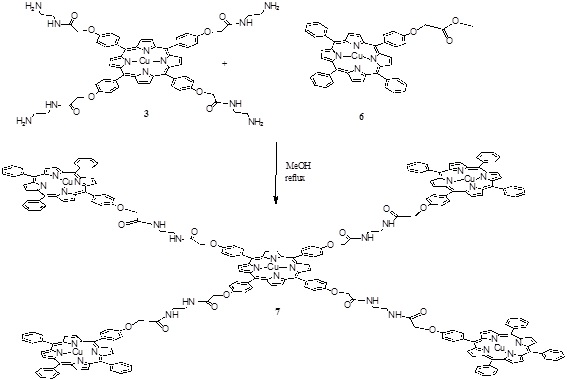 |
Scheme (3) Synthesis of the penta porphyrin array. |
The pentamer was obtained in one step by an amidation reaction between the compounds 3 and 6 (Scheme 3 ) in methanol at reflux for 4 hours giving compound 7 with a 35 % yield.
) in methanol at reflux for 4 hours giving compound 7 with a 35 % yield.
The 1H-NMR spectra of compound 7 exhibits: two signals at δH 1.60 and at δH 2.95 for the N-H and ethyl protons, signals at δH 4,80 and 4.94 for the Ar-CH2-O, and for the protons of the aromatic substituents seven signals at δH 7.08, 7.50, 7.66, 7.78, 7.98, 8.23, and at 8.59. And the beta pyrrolic protons at δH 8.83 were observed. The full functionalization of the porphyrin was corroborated by MALDI-TOF and is observed at 4064 m/z the mass of compound 7 (Fig. 1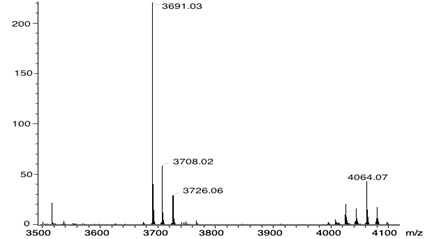 ).
).
 |
Fig. (1) MALDI-TOF mass spectra of the compound 7. |
2.1. Electron Paramagnetic Resonance (EPR) Studies
Porphyrins have delocalized π electrons. The EPR signals of these delocalized π electron systems are generated by the interaction of the porphyrin, or in the case of metal-porphyrins, by the unpaired electrons of a metal ion with the magnetic field (Fig. 2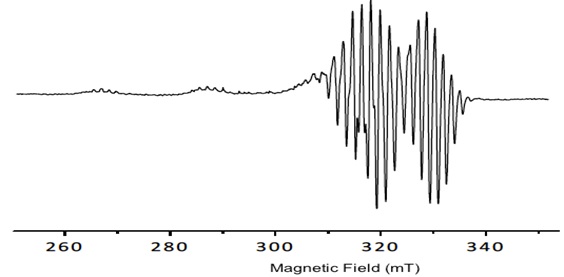 ).
).
 |
Fig. (2) Spectral behavior of the porphyrin 1. |
The copper porphyrin complex exhibits an anisotropic EPR spectra typical of copper (II) centers with well determined hyperfine lines in the gǁ. All signals show the dependence gǁǁ> g┴ >ge which is diagnostic of a dx2-y2 ground state for copper (II), the hyperfine structure is obtained from the contact between delocalized π electrons and the nuclear spin ICu=3/2. The values obtained by simulated spectra are shown in Table 1. Additionally, in the spectra, a super hyperfine structure can be observed due to the coupling of with the nuclei of pyrrole nitrogens of Cu-porphyrin IN =1 (shfcc). The simulated spectra show the values in Table 1.
The tetrahedral distortion that frequently occurs for square planar copper complexes is observed by the ratio f = gǁǁ/Aǁǁ and it is considered for the determination of the distortion and the deviation from perfect geometry depending on the coordinated ligands [26Labanova, M.; Bidzinska, E.; Para, A. EPR investigation of Cu(II)-complexes with nitrogen derivatives of dialdehyde starch. Carbohydr. Polym., 2012, 87, 2605-2613.
[http://dx.doi.org/10.1016/j.carbpol.2011.11.034] , 27Pogni, R.; Baratto, M.C.; Diaz, A.; Basosi, R.J. Inorg. Biochemistry, 2000, 79, 333-339.
[http://dx.doi.org/10.1016/S0162-0134(99)00166-X] ]. The f value in the range 110-120 is typical for signals of the copper complex with planar geometry, however, its increase to 150 indicates a small or modest distortion of the planar symmetry, and the higher values, 180- 250, suggest a strong deviation from square planar geometry. In general, as a complex becomes more tetrahedral, the gǁ value and the giso value increase and the Aiso and Aǁǁ values decrease [28Cline, S.J.; Wasson, J.R.; Hatfield, W.E.; Hodgson, D.J. Structure and spectroscopic properties of bis(N-cyclohexyl-3-methoxysalicylideneiminato)-copper(II). J. Chem. Soc., Dalton Trans., 1978, 1051-1057.
[http://dx.doi.org/10.1039/dt9780001051] ]. Additionally, the EPR spectra can help with an estimation of the geometry that the compounds have adopted, by using the isotropic parameters Aiso= (A|| +2A┴ )/3 and giso= (g|| + 2g┴ )/3. The values for planar symmetry are between Aiso= 75 -90 cm-1 and giso= 2.06 - 2.11 [29Suresh, E.; Bhadbhade, M.M.; Srinivas, D. Molecular association, chelate conformation and reactivity correlations in substituted o-phenylenebis (salicylidenato) copper(II) complexes: UV-visible, EPR and X-ray structural investigations. Polyhedron, 1996, 15, 4133-4144.
[http://dx.doi.org/10.1016/0277-5387(96)00178-7] , 30Joseph, M.; Kuriakose, M.; Kurup, M.R.; Suresh, E.; Kishore, A.; Bath, S.G. Structural, antimicrobial and spectral studies of copper(II) complexes of 2-benzoylpyridine N(4)-phenyl thiosemicarbazone. Polyhedron, 2006, 25, 61-70.
[http://dx.doi.org/10.1016/j.poly.2005.07.006] ].

The results show no variation in the tetrahedral distortion (Table 1) among all compounds, since all the values are close to f = 106 cm-1 indicating a planar geometry. No increase in the tetrahedral distortion of the porphyrin ring was observed because of ring stiffness. The values found for giso (2.09 to 2.10) and Aiso 75 to 85 cm-1 are consistent with a square planar geometry.
2.2. Photophysical Studies in Solution
The absorption spectra of the metalloporphyrins 1, 2, 3, 5, 6 and 7 showed similar Soret or B and Q bands (Fig. 3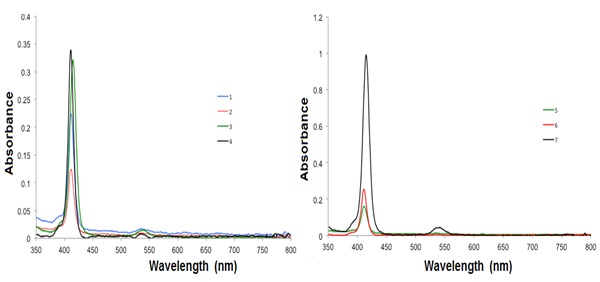 ). A small ipsochromic change of the B band by a some nm can be perceived in the pentamer porphyrin array when compared to the monomeric porphyrin Cu(II), which signifies a faint excitonic coupling amid the subunits in the porphyrin, independent of the concentration (1 x 10−6 M−1 and 3 x 10−8 M−1). The porphyrins are quite undisturbed and the porphyrins are alone mild electronically coupled [31Holten, D.; Bocian, D.F.; Lindsey, J.S. Probing electronic communication in covalently linked multiporphyrin arrays. A guide to the rational design of molecular photonic devices. Acc. Chem. Res., 2002, 35(1), 57-69.
). A small ipsochromic change of the B band by a some nm can be perceived in the pentamer porphyrin array when compared to the monomeric porphyrin Cu(II), which signifies a faint excitonic coupling amid the subunits in the porphyrin, independent of the concentration (1 x 10−6 M−1 and 3 x 10−8 M−1). The porphyrins are quite undisturbed and the porphyrins are alone mild electronically coupled [31Holten, D.; Bocian, D.F.; Lindsey, J.S. Probing electronic communication in covalently linked multiporphyrin arrays. A guide to the rational design of molecular photonic devices. Acc. Chem. Res., 2002, 35(1), 57-69.
[http://dx.doi.org/10.1021/ar970264z] [PMID: 11790089] ].
The difference in the absorption intensity of the porphyrin-core derivatives 1-6 in comparison with the pentaporphyrin array is caused by the porphyrin substituents on the porphyrin-connected phenyl rings. As the porphyrin derivative size intensities, no shifts in the fluorescence and absorption bands were observed, indicating that the substituents, which are targeted outward from the porphyrin center, do not engage with the dyes [32Falk, J.E. Porphyrins and Metalloporphyrins; Elsevier: New York, 1964, pp. 85-87.]. Therefore, the substituents are not assumed to change either the character of the photoexcitation or the consequent photophysical processes of the porphyrin moiety.
When excited at 425 nm, all Cu-porphyrins emit at 605 nm and at 659 nm (Fig. 4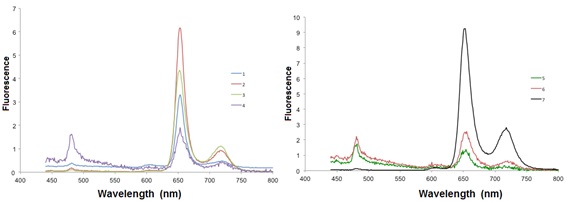 ).
).
Table 2 shows the absorption spectral data of compounds 1-7 in CH2Cl2 at 24 °C, where the molar extinction coefficient (ε) of the absorbance of the compounds 1-7 showed an increase for the compounds 2-4, and decrease for the compounds 5-7, while the quantum yield showed slightly changes.
2.3. Morphological Film Properties
In Fig. (5 ), the surface morphology of the film from compound 7 is shown. From this image, a very regular distribution is observed. In addition, the surface coverage is complete which suggests a homogenous deposit on the ITO substrate.
), the surface morphology of the film from compound 7 is shown. From this image, a very regular distribution is observed. In addition, the surface coverage is complete which suggests a homogenous deposit on the ITO substrate.
The surface morphology was further analyzed using atomic force microscopy. In Fig. (6 ), an AFM image of compound 7, is observed, which shows a granular aspect. From this image, it is clear that the surface coverage is not homogeneous, since small defects or holes can be seen irregularly distributed on the film surface. The thickness of the film was 30nm and the mean roughness value was 1.33 nm, which nevertheless, suggests the presence of a very flat film.
), an AFM image of compound 7, is observed, which shows a granular aspect. From this image, it is clear that the surface coverage is not homogeneous, since small defects or holes can be seen irregularly distributed on the film surface. The thickness of the film was 30nm and the mean roughness value was 1.33 nm, which nevertheless, suggests the presence of a very flat film.
2.4. Conductivity Studies
In addition to the morphological characterization, the surface conductivity of the film of 7 was measured. In Fig. (7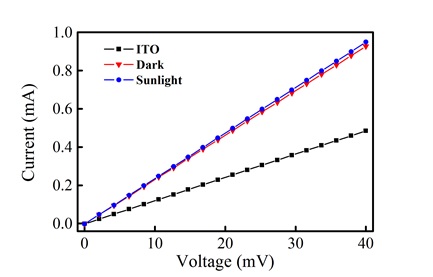 ), a conductivity (V vs. I) plot of the bare ITO substrate and film in the presence and absence of light is shown. From these curves, the linear resistance of the bare ITO was calculated as 82.17 ohms, while the film shows a linear resistance of 43.05 ohms. This change suggests that the film exhibits a larger conductivity in comparison to the ITO substrate. In the presence of light, there is a slight decrease in linear resistance (42.02 ohms), which suggests a very small photoconductive response.
), a conductivity (V vs. I) plot of the bare ITO substrate and film in the presence and absence of light is shown. From these curves, the linear resistance of the bare ITO was calculated as 82.17 ohms, while the film shows a linear resistance of 43.05 ohms. This change suggests that the film exhibits a larger conductivity in comparison to the ITO substrate. In the presence of light, there is a slight decrease in linear resistance (42.02 ohms), which suggests a very small photoconductive response.
 |
Fig. (3) Absorption spectra of the compounds 1-7 in CH2Cl2 at room temperature. |
3. CONCLUSION
A new class of porphyrin array has been synthesized. The Cu(II) porphyrin-PAMAM-Cu(II) porphyrin was obtained in good yields. The EPR studies showed that all porphyrins obtained have signals of a copper complex with planar geometry, and no variation in the tetrahedral distortion of the porphyrin ring was observed. The UV-vis and emission studies showed that the intensity of the signal was increased due to the presence of five porphyrins in the structure. On the other hand, the Cu(II) porphyrin-PAMAM-Cu(II) porphyrin produced regular films when deposited onto ITO surfaces. The morphology showed a granular aspect with few scattered defects or vacancies. The photoconductive response of the film was not very large, possibly due to the irregularities on the film surface, which might affect the conduction paths or inefficient molecular packing due to the molecular geometry. Nevertheless, the conductivity of the film is larger than the one measured for the bare ITO surface, which suggests the presence of a conductive organic film.
4. EXPERIMENTAL SECTION
4.1. Materials and Equipment
Solvents and reagents were purchased as reagent grade. 1H and 13C NMR spectra were recorded on a Varian Unity-300 MHz in CDCl3. The UV-Vis absorption and emission spectra were performed using a Shimadzu 2401 PC spectrophotometer and Perkin-Elmer LS-50 spectrofluorimeter. Quantum yields (ΦF) were determined using a quinine solution in CH2Cl2 as fluorescence standard (ΦF (std) = 0.546). All compounds were dissolved in dichloromethane (n = 1.445) and the fluorescence standard (nstd= 1.465). The EPR measurements were made in a Jeol JES-TE300 spectrometer. The g- factor values were calculated according to Weil [33Weil, J.A.; Bolton, J.R. Electron Paramagnetic Resonance; Wiley Interscience: New York, 2007. ]. The films for the photoelectric measurements were prepared on glass plates covered with a semitransparent layer of indium tin oxide (ITO) with a sheet resistance of 10 ohms/sq. The substrates were cleaned and nitrogen was dried before each evaporation. The Cu(II) porphyrin-PAMAM-Cu(II) porphyrin compound was deposited by using a NORM vacuum thermal evaporator VCM 600 working at a 10-6 mbar pressure. The morphological properties of the film were evaluated with a JEOL FE7800F scanning electron microscope (SEM) working at 1kV in the gentle beam mode, in order to minimize the destruction and charging of the films. In addition, atomic force microscopy (AFM) images were obtained with a JEOL JSPM4210 instrument in the tapping mode to determine the morphology, thickness and roughness of the films.
 |
Fig. (4) Emission spectra of the compounds 1-7 in CH2Cl2 at room temperature. |
The photoconductive response of the film was evaluated with a Keithley 2400 digital SourceMeter instrument and an Oriel LCS-100 solar simulator lamp with an AM1.5G filter.
 |
Fig. (5) SEM image of the metalloporphyrin film. Image magnification is 1,000X. |
4.2. Synthesis
Copper porphyrin 1
5,10,15,20-tetrakis(4-hydroxyphenyl)-21H,23H-porphine 200 mg (0.294 mmol) and Cu(OAc)2 58 mg (0.294 mmol) were dissolved in methanol (20 mL), and refluxed for 4 h, then distilled water (60 mL) was added, and the methanol was evaporated under vacuum. The solution was cooled, and the purple crystalline product was obtained (0.28 mmol), FTIR (pellet, KBr, cm-1): 3245, 1606, 1502, 1431, 1342, 1209, 1168, 999, 794, 719, 532. UV-vis (CH2Cl2): λmax 417, 540 nm. 1H NMR (300 MHz, in CDCl3) δH: 6.62 (t, 4 H, Ar-H), 7.01 (d, 8 H, Ar-H), 8.86 (s, 8 H, Ar-H), 9.60 (s, 8 H, Ar-OH). 13C NMR (75 MHz, in CDCl3) δC: 101.72 (Ar-H). 114.16 (Ar), 120.23 (Ar), 131.33 (Ar), 156.21 (Ar-OH), 144.46 (Ar-porph), 148.93 (Ar), MS (FAB+) m/z: 739 (M+). Anal. Calcd for C44H28CuN4O4. C 71.39 %, H 3.81 %, Cu 8.58 %, N 7.57 %, O 8.65 %. Found: C, 71.40, H, 3.83, N, 7.57 %.
Compound 2
Methyl bromoacetate 22.8 mg (1.60 mmol) and potassium carbonate (3.20 mmol) in dry acetone (50 mL) were refluxed for 20 min. The PcCu(II) 1, 122 mg (1.60 mmol) in dry acetone (40 mL) was added drop wise and stirred for 6 hours. The mixture was filtered. The filtrate was evaporated to dryness under pressure. The residue dissolved in diethyl ether and washed with a solution of 5% Na2CO3 in H2O (3 times). The organic layer was evaporated to give the ester 2 (0.27 mmol) as a purple solid in 90 % yield. m.p: ˃ 300 °C. FTIR (pellet, KBr, cm-1): 2954, 2920, 1750, 1602, 1503, 1430, 1298, 1207, 1173, 1140, 1076, 963, 806, 708, 604, 593. UV-vis (CH2Cl2): λmax 417, 540. 1H NMR (300 MHz, CDCl3) δH: 3.89 (br, 12H, O-CH3), 4.82 (br, 8H, CH2-O), 7.06 (br, 24H, Ar, pyrrol). 13C NMR (75 MHz, CDCl3) δC: 52.4 (CH3-O), 65.4 (CH2-O), 112.5 (Ar), 119.4 (Aripso), 137.2 (pyrrol), 141.0 (Ar), 145.7 (pyrrolipso), 157.1 (Aripso-O), 169.4 (C=O). MS (FAB+) m/z: 1027 (M+). Anal. Calcd for C56H44CuN4O12. C 65.40 %, H 4.31 %, Cu 6.18 %, N 5.45 %, O 19.85 %. Found: C, 65.43, H, 4.3, N, 5.42 %.
 |
Fig. (6) AFM image of the metalloporphyrin film. Image size is 2 x 2 µm. |
 |
Fig. (7) V vs. I plots for the bare ITO surface and the porphyrin thin film in the presence and absence of light. |
Compound 3
A mixture of ester-containing derivative 2 190 mg (0.197 mmol) and ethylenediamine 81 mg (0.788 mmol) in methanol: toluene 1:1 was stirred and refluxed for 24 h. The solvents were removed in vacuo. The residue was washed several times with alcohol to obtain the amide compound 3 (0.16 mmol) as a purple solid in 85 % yield. m.p: ˃ 300 °C. FTIR (pellet, KBr, cm-1): 3302, 3032, 2917, 1655, 1602, 1533, 1498, 1343, 1284, 1225, 1174, 996, 799, 536. UV-vis (CH2Cl2): λmax 417, 541, 682. 1H NMR (300 MHz, CDCl3) δH: 1.60 (br, 8H, NH2), 3.90 (m, 16H, CH2-NH2), 4.83 (s, 8H, CH2-O), 7.06 (br, 24H, Ar, pyrrol). 13C NMR (75 MHz, CDCl3) δC: 52.4 (CH2-NH2), 65.5 (CH2-O) 112.5 (Ar), 115.0 (Aripso), 137.0 (pyrrol), 157.1 (Ar-O), 169.4 (C=O). MS (FAB+) m/z: 1140 (M+). Anal. Calcd for C60H60CuN12O8. C 63.17 %, H 5.30 %, Cu 5.57 %, N 14.73 %. Found: C, 63.19; H, 5.29, N 14.72 %.
5-(4-Hydroxyphenyl)-5,10,15-triphenyl-porphyrin 4
Benzaldehyde 2.08 g (0.02 mmol),) para-hydroxybenzaldehyde 2.45 g (0.02 mmol), and pyrrole 2.72 g (0.04 mmol) were added in propionic acid (15 mL) for 3 h. After the workup the reaction mixture was washed with water and chromatographed on a silica gel column using a mixture of hexane-ethyl acetate. Obtaining 4 0.23 g as a purple solid in 37 % yield. m.p: ˃ 300 °C. FTIR (pellet, KBr, cm-1): 3503, 3331, 2919, 1606, 1559, 1469, 1167, 793. UV-vis (CH2Cl2): λmax 408, 515, 550, 590, 646. 1H NMR (300 MHz, CDCl3) δH: -2.79 (br, 2H, NH), 7.24 (d, 2H, Ar, J = 9.0 Hz), 8.16 (m, 6H, Ar), 8.09 (d, 2H, Ar, J= 9.0 Hz), 8.21-8.24 (m, 3H, Ar), 8.85 (m, 6H, Ar), 8.88 (br, 8H, pyrrol). 13C NMR (75 MHz, CDCl3) δC: 113.7 (Ar), 120.0 (Ar-porph), 126.0 (Ar), 127.7 (Ar), 131.0 (pyrrol), 134.5 (Ar), 140.3 (Ar), 142.2 (pyrrolipso), 143.2 (Ar-O). MS (FAB+) m/z: 630 (M+). Anal. Calcd for C44H30N4O. C 83.79 %, H 4.79 %, N 8.88 %. Found: C, 83.77; H, 4.79, N 8.86 %.
Compound 5
A mixture of 4 0. 1 g (0.158 mmol) and Cu(OAc )2 0.1 g (0.23 mmol) in methanol (20 mL), was heated to reflux for 4 h. Following the same methodology reported above, giving 5 0.150 g (0.21 mmol, yield 93 %). m.p: ˃ 300 °C. FTIR (pellet, KBr, cm-1): 3317, 2919, 1759, 1739, 1597, 1437, 1218, 1177, 795, 700. UV-vis (CH2Cl2): λmax 408, 515, 550, 590, 646. 1H NMR (300 MHz, CDCl3) δH: 7.52 (br, 2H, Ar), 7.75 (br, 6H, Ar), 8.08 (br 2H, Ar), 8.21 (br, 3H, Ar), 8.87 (br, 6H, Ar), 8.88 (br, 8H, pyrrol). 13C NMR (75 MHz, CDCl3) δC: 113.7 (Ar), 120.0 (Ar-porph), 126.0 (Ar), 127.7 (Ar), 130.8 (pyrrol), 134.5 (Ar), 135.7 (Ar), 142.2 (Ar-O). MS (FAB+) m/z: 692 (M+). Anal. Calcd for C44H28CuN4O. C 76.34 %, H 4.08 %, N 8.09 %. Found: C, 76.32; H, 4.09, N 8.06 %.
Compound 6
Methyl bromoacetate 0.241 g (1.60 mmol), potassium carbonate (3.20 mmol) dissolved in dry acetone (50 mL) and heated to reflux and stirred vigorously in a nitrogen atmosphere for 20 min. The compound 5 0.110 g (1.60 mmol) dissolved in dry acetone (40 mL) was added drop wise and the reaction was continued for 6 hours. The mixture was filtered. The filtrate was dried under pressure. The monoester 6 was obtained following the same methodology used for the compound 3. Obtaining 0.206 g (0.27 mmol) as a purple solid in 90 % yield. m.p: ˃ 300 °C. FTIR (pellet, KBr, cm-1): 3034, 2924, 1760, 1501, 1270, 1175, 1070, 996, 805, 702. UV-vis (CH2Cl2): λmax 418, 540. 1H NMR (300 MHz, CDCl3) δH: 3.74 (s, 3H, O-CH3), 4.22 (s, 2H, CH2-O), 7.27 (br, 2H, Ar), 7.73 (br, 6H, Ar), 8.13 (br, 2H, Ar), 8.21 (br, 3H, Ar), 8.83 (br, 14H, Ar, pyrrol).13C NMR (75 MHz, CDCl3) δC: 52.3 (O-CH3), 68.10 (CH2-O), 112.9 (Ar), 120.1 (Ar-porph), 126.6 (Ar), 127.1 (Aripso) 127.7 (Ar), 130.8 (pyrrol), 134.5 (Ar), 135.6 (Ar), 142.2 (Ar-O), 170.0 (C=O). MS (FAB+) m/z: 763 (M+). Anal. Calcd for C47H32CuN4O3. C 73.86 %, H 4.22 %, N 7.33 %. Found: C, 73.84; H, 4.23, N 7.31 %.
Compound 7
The compound 6 100 mg (0.144 mmol) and the amide compound 3 114 mg (0.144 mmol) were dissolved in methanol (20 mL). The mixture was heated to reflux for 4 h, and then the methanol was evaporated under vacuum. The purple crystalline product was dried in vacuo at 40 °C (yield 35 %). m.p: ˃ 300 °C. FTIR (pellet, KBr, cm-1): 3033, 2920, 2850, 1727, 1600, 1499, 1213, 1174, 1071, 798, 703. UV-vis (CH2Cl2): λmax 413, 449, 540, 622, 776. 1H NMR (300 MHz, CDCl3) δH: 1.60 (br, 8H, NH), 2.95 (m, 16H, CH2-NH), 4.80 (s, 8H, CH2-O), 4.94 (s, 8H, CH2-O), 7.08 (br, 8H, Ar), 7.50 (br, 8H, Ar), 7.66 (br, 24H, Ar), 7.78 (br, 8H, Ar), 7.98 (br, 8H, Ar), 8.23 (br, 12H, Ar), 8.59 (br, 24H, Ar), 8.83 (br, 40H, pyrrol). 13C NMR (75 MHz, CDCl3) δC: 49.1 (CH2-NH2), 65.2 (CH2-O) 112.5 (Ar), 115.0 (Aripso), 126.3 (Ar), 126.7 (Ar), 127.0 (Ar), 128.2 (Ar), 137.0 (pyrrol), 157.1 (Ar-O), 171.0 (C=O). MALDI-TOF m/z: 4064 (M+). Anal. Calcd for C244H172Cu5N28O16. C 72.01 %, H 4.26 %, N 9.64 %. Found: C, 72.00; H, 4.28, N 9.63 %.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This work was supported by the DGAPA-UNAM (IN-107814) grant. MR acknowledges DGAPA-UNAM IN-106513 grant. O. Amelines-Sarria acknowledges financial support from DGAPA CJIC/CTIC/1334/2013. We would also like to thank Rios O. H., Velasco L., Huerta S. E., Patiño M. M. R., Peña Gonzalez M. A., Garcia Rios E. and Magaña Zavala C.R. for technical assistance.
REFERENCES
| [1] | Chen, C.T. In Comprehensive Supramolecular Chemistry: Coordination Polymers; Pergamon Press: New York, 1996, vol. 5, no. 4, p. 91. |
| [2] | Sanders, J.K. The Porphyrin Handbook; Kadish, K.M.; Smith, K.M.; Guilard, R., Eds.; Academic Press: New York, 2000, vol. 3, p. 347. |
| [3] | Ward, M.D. Photo-induced electron and energy transfer in non-covalently bonded supramolecular assemblies. Chem. Soc. Rev., 1997, 26, 365-375. [http://dx.doi.org/10.1039/cs9972600365] |
| [4] | Xu, Z.; Moore, J.S. Design and synthesis of a convergent and directional molecular antenna. Acta Polym., 1994, 45, 83-87. [http://dx.doi.org/10.1002/actp.1994.010450204] |
| [5] | Drain, C.M.; Lehn, J.M. Self-assembly of square multiporphyrin arrays by metal ion coordination. J. Chem. Soc. Chem. Commun., 1994, 24, 2313-2315. [http://dx.doi.org/10.1039/c39940002313] |
| [6] | Chi, X.; Guerin, A.J.; Haycock, R.A.; Hunter, C.A.; Sarson, L.D. The thermodynamics of self-assembly. J. Chem. Soc. Chem. Commun., 1995, 2567-2569. [http://dx.doi.org/10.1039/c39950002567] |
| [7] | Miyaji, H.; Kobuke, Y.; Kondo, J. Molecular organization of mono(p-hydroquinonyl) porphyrin through coordinate bond. Chem. Lett., 1996, 6, 497-498. [http://dx.doi.org/10.1246/cl.1996.497] |
| [8] | Tamiaki, H.; Amakawa, M.; Shimono, Y.; Tanikaga, R.; Holzwarth, A.R.; Schaffner, K. Synthetic zinc and magnesium chlorin aggregates as models for supramolecular antenna complexes in chlorosomes of green photosynthetic bacteria. Photochem. Photobiol., 1996, 63, 92-99. [http://dx.doi.org/10.1111/j.1751-1097.1996.tb02997.x] |
| [9] | Alessio, E.; Macchi, M.; Heath, S.; Marzilli, L. G. A novel open-box shaped pentamer of vertically linked porphyrins that selectively recognizes S-bonded Me2SO complexes. Chem. Commun. (Camb.), 1996, 1411-1412. [http://dx.doi.org/10.1039/cc9960001411] |
| [10] | Kariya, N.; Imamura, T.; Sasaki, Y. Synthesis, characterization, and spectral properties of new perpendicularly linked osmium(II) porphyrin oligomers1. Inorg. Chem., 1997, 36, 833-839. [http://dx.doi.org/10.1021/ic960878k] |
| [11] | Denti, G.; Serroni, S.; Campagna, S.; Juris, A.; Balzani, V. Small-upward approach to nanostructures-dendritic polynuclear metal-complexes for light-harvesting. Mol. Cryst. Liq. Cryst. (Phila. Pa.), 1993, 234, 79-88. [http://dx.doi.org/10.1080/10587259308042900] |
| [12] | Bothner-By, A.A.; Dadok, J.; Johnson, T.E.; Lindsey, J.S. Molecular dynamics of covalently-linked multi-porphyrin arrays. J. Phys. Chem., 1996, 100, 17551-17557. [http://dx.doi.org/10.1021/jp961408e] |
| [13] | Wagner, R.W.; Lindsey, J.S. A molecular photonic wire. J. Am. Chem. Soc., 1994, 116, 9759-9760. [http://dx.doi.org/10.1021/ja00100a055] |
| [14] | Anderson, S.; Anderson, H.L.; Bashall, A.; McPartlin, M.; Sanders, J.K. Assembly and crystal structure of a photoactive array of five porphyrins. Angew. Chem. Int. Ed. Engl., 1995, 34, 1096-1099. [http://dx.doi.org/10.1002/anie.199510961] |
| [15] | Drain, C.M.; Russell, K.C.; Lehn, J.M. Self-assembly of a multi-porphyrin supramolecular macrocycle by hydrogen bond molecular recognition. Chem. Soc. Chem. Commun. (Camb.), 1996, 337-338. [http://dx.doi.org/10.1039/cc9960000337] |
| [16] | Huck, W.T.; Rohrer, A.; Anilkumar, A.T.; Fokkens, R.H.; Nibbering, N.M.; van Veggel, F.C.; Reinhoudt, D.N. Non-covalent synthesis of multiporphyrin systems. New J. Chem., 1998, 22, 165-168. [http://dx.doi.org/10.1039/a708323h] |
| [17] | Drain, C.M.; Nifiatis, F.; Vasenko, A.; Batteas, J.D. Porphyrin tessellation by design: metal-mediated self-assembly of large arrays and tapes. Angew. Chem. Int. Ed., 1998, 37, 2344-2347. [http://dx.doi.org/10.1002/(SICI)1521-3773(19980918)37:17<2344::AID-ANIE2344>3.0.CO;2-B] |
| [18] | Kumar, R.K.; Goldberg, I. Supramolecular assembly of heterogeneous multiporphyrin arrays-structures of [{ZnII(tpp)}2(tpyp)] and the coordination polymer [{[MnIII(tpp)]2(tpyp)(ClO4)2}∞]. Angew. Chem. Int. Ed., 1998, 37, 3027-3030. [http://dx.doi.org/10.1002/(SICI)1521-3773(19981116)37:21<3027::AID-ANIE3027>3.0.CO;2-N] |
| [19] | Taylor, P.N.; Anderson, H.L. Cooperative self-assembly of double-strand conjugated porphyrin ladders. J. Am. Chem. Soc., 1999, 121, 11538-11548. [http://dx.doi.org/10.1021/ja992821d] |
| [20] | Solladié, N.; Gross, M.; Gisselbrecht, J.P.; Sooambar, C. Pentaporphyrin with flexible, chiral nucleosidic linkers: unexpected duality of the physico-chemical properties of its core. Chem. Commun. (Camb.), 2001, 21(21), 2206-2207. [http://dx.doi.org/10.1039/b106337p] [PMID: 12240113] |
| [21] | Flamigni, L.; Talarico, A.M.; Ventura, B.; Marconi, G.; Sooambar, C.; Solladié, N. Conformational effects on the photoinduced energy transfer in a star-shaped pentaporphyrin with nucleosidic linkers. Eur. J. Inorg. Chem., 2004, 12, 2557-2569. [http://dx.doi.org/10.1002/ejic.200300918] |
| [22] | Solladié, N.; Sooambar, C.; Herschbach, H.; Strub, J.M.; Leize, E.; Van Dorsselaer, A.; Talarico, A.M.; Ventura, B.; Flamigni, L. A photoactive nona-porphyrin with nucleosidic linkers. New J. Chem., 2005, 29, 1504-1507. [http://dx.doi.org/10.1039/b509913g] |
| [23] | Wagner, R.W.; Lindsey, J.S.; Seth, J.; Palaniappan, V.; Bocian, D.F. Molecular optoelectronic gates. J. Am. Chem. Soc., 1996, 118, 3996-3997. [http://dx.doi.org/10.1021/ja9602657] |
| [24] | James, D.A.; Arnold, D.P.; Parsons, P.G. Potency and selective toxicity of tetra(hydroxyphenyl)- and tetrakis(dihydroxyphenyl)porphyrins in human melanoma cells, with and without exposure to red light. Photochem. Photobiol., 1994, 59(4), 441-447. [http://dx.doi.org/10.1111/j.1751-1097.1994.tb05062.x] [PMID: 8022886] |
| [25] | Vaijanthimala, G.; Krishnan, V.; Mandal, S.K. Cyclic porphyrin dimers as hosts for coordinating ligands. J. Chem. Sci., 2008, 120, 115-129. [http://dx.doi.org/10.1007/s12039-008-0014-3] |
| [26] | Labanova, M.; Bidzinska, E.; Para, A. EPR investigation of Cu(II)-complexes with nitrogen derivatives of dialdehyde starch. Carbohydr. Polym., 2012, 87, 2605-2613. [http://dx.doi.org/10.1016/j.carbpol.2011.11.034] |
| [27] | Pogni, R.; Baratto, M.C.; Diaz, A.; Basosi, R.J. Inorg. Biochemistry, 2000, 79, 333-339. [http://dx.doi.org/10.1016/S0162-0134(99)00166-X] |
| [28] | Cline, S.J.; Wasson, J.R.; Hatfield, W.E.; Hodgson, D.J. Structure and spectroscopic properties of bis(N-cyclohexyl-3-methoxysalicylideneiminato)-copper(II). J. Chem. Soc., Dalton Trans., 1978, 1051-1057. [http://dx.doi.org/10.1039/dt9780001051] |
| [29] | Suresh, E.; Bhadbhade, M.M.; Srinivas, D. Molecular association, chelate conformation and reactivity correlations in substituted o-phenylenebis (salicylidenato) copper(II) complexes: UV-visible, EPR and X-ray structural investigations. Polyhedron, 1996, 15, 4133-4144. [http://dx.doi.org/10.1016/0277-5387(96)00178-7] |
| [30] | Joseph, M.; Kuriakose, M.; Kurup, M.R.; Suresh, E.; Kishore, A.; Bath, S.G. Structural, antimicrobial and spectral studies of copper(II) complexes of 2-benzoylpyridine N(4)-phenyl thiosemicarbazone. Polyhedron, 2006, 25, 61-70. [http://dx.doi.org/10.1016/j.poly.2005.07.006] |
| [31] | Holten, D.; Bocian, D.F.; Lindsey, J.S. Probing electronic communication in covalently linked multiporphyrin arrays. A guide to the rational design of molecular photonic devices. Acc. Chem. Res., 2002, 35(1), 57-69. [http://dx.doi.org/10.1021/ar970264z] [PMID: 11790089] |
| [32] | Falk, J.E. Porphyrins and Metalloporphyrins; Elsevier: New York, 1964, pp. 85-87. |
| [33] | Weil, J.A.; Bolton, J.R. Electron Paramagnetic Resonance; Wiley Interscience: New York, 2007. |