- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Open Chemistry Journal
(Discontinued)
ISSN: 1874-8422 ― Volume 8, 2021
A New Synthetic Method of 1,5-Dimethyl-3-Oxabicyclo[3.1.0] Hexane-2,4-Dione
Youcef Mechehoud1, Nadjah Belattar1, *, Samir Benayache1, Fadila Benayache1, Paul Mosset2
Abstract
Background:
We have described in this reported work a new method in the synthesis of cis-1,5-dimethyl -3-oxabicyclo [3.1.0] hexane-2,4-dione in good yield.
Objective:
Optimization of practical conditions leads to obtain 1,3-cyclopropanedicarboxylic anhydrides as important precursors of functionalized cyclopropane derivatives.
Method:
The condensation of 2-chloropropanoic acid with ethyl methacrylate using (2M) LDA dissolved in hexane and THF at (-80°C), and the treatment with acetyl chloride permit to obtain the substituted 1,3- cyclopropanedicarboxylic anhydride .
Results:
We have proceeded to the synthesis of cis-1,5-dimethyl -3-oxabicyclo [3.1.0] hexane-2,4-dione as functionalized organic compound with high efficiency ,taking into account the regioselectivity of carbanion attack to double bond activated by an electrophilic group.
Conclusion:
Using (2M) LDA dissolved in hexane and THF at (-80°C) is a good way to afford the enantioselective substituted 1,3- cyclopropanedicarboxylic anhydrides.
Article Information

Identifiers and Pagination:
Year: 2018Volume: 5
First Page: 44
Last Page: 50
Publisher Id: CHEM-5-44
DOI: 10.2174/1874842201805010044
Article History:
Received Date: 09/03/2018Revision Received Date: 10/05/2018
Acceptance Date: 31/05/2018
Electronic publication date: 29/06/2018
Collection year: 2018
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: https://creativecommons.org/licenses/by/4.0/legalcode. This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
* Address correspondence to this author at the Research Unity of Valorization of Natural Resources, Bioactive Molecules and Physicochemical Biological Analysis, Department of Chemistry, Faculty of Exact Sciences, University of Mentouri- Brothers, Constantine-1, Algeria; E-mail: nadjahorg@gmail.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 09-03-2018 |
Original Manuscript | A New Synthetic Method of 1,5-Dimethyl-3-Oxabicyclo[3.1.0] Hexane-2,4-Dione | |
1. INTRODUCTION
The natural cyclopropanes, generated ephemerally in primary and secondary metabolisms and the synthetic cyclopropanes, carrying simple functionalities are endowed with a large spectrum of biological properties ranging from enzyme inhibitions to antimicrobial, antiviral, antitumor and neurochemical properties [1Salaün, J.; Baird, M.S. Curr. Med. Chem., 1995, 2, 511.], they provide convenient biological probes for mechanistic studies and allow the production of new drugs [2Liu, H.W.; Walsh, C.T. Biochemistry of the Cyclopropyl Group, 1987, Chap 16; Kennewell PDbMatharu, S.S. J. Chem.Soc .Perkin Trans, 1982, 1, 2553.]. Moreover, some synthetic derivatives such as (S) bioallethrin or bioresmethrin present a high insecticidal activity with low mammalian toxicity [3 a) M.Eliott . Synthetic pyrethroids ,ACS .Symposium Series ,No.42: Washington DC,1977. b) M.Eliott.; N.F.James .Chem.Soc.Rev, 1978, 7,473 . c) Imperial Chemical Industries Ltd.Brit, 1,537,449; Chem Abstr, 1979,91,51102 w.].
The cyclopropane compounds and their derivatives have been synthesized via Michael addition taking into account their asymmetric synthesis and more particularly their stereochemistry dependence of solvent polarity .These processes are initiated by nucleophilic addition of carbanions to acrylates, followed by the 1,3-eliminative cyclization. It's about the base-catalyzed condensation of α-haloesters with α,β-unsaturated esters [4 a) McCoy, L.L. J. Am. Chem. Soc., 1958, 80, 6568. b) McCoy, L.L. J. Am. Chem. Soc., 1962, 84, 2246. c) McCoy, L.L. J. Org. Chem., 1960, 25,
2078.Fraisse, R. Bull; Soc.Chem: France, 1959, p. 1102.].The striking solvent polarity –dependence of steric orientation in their asymmetric synthesis has been elaborated by Inouye and his collaborators [5 a) Inouye, Y.; Horiike., Ms; Inouye, Y.; Inouye, Y.; Inouye, Y. J. Am. Chem. Soc., 1961, 83, 2962. b) Inouye, Y. Tetrahedron, 1968, 24, 2907.
[http://dx.doi.org/10.1021/ja01474a048] [http://dx.doi.org/10.1016/S0040-4020(01)98699-5] ].
Furthermore, Schmidt [6Schmidt, U. R.Schröer and A.Hochrainer. Liebigs Ann., 1970, 773, 180.
[http://dx.doi.org/10.1002/jlac.19707330120] ] suggested a mechanism involving the initial nucleophilic attack of carbanion to the polarized double bond of the Michael acceptor, followed by the probable 1,3-shift of proton in the intermediate adduct carbanion and then the intramolecular nucleophilic displacement leads to cyclopropane products.
Our work has been based on the preparation of cyclopropanedicarboxylic anhydrides, which are the important precursors of various functionalized cyclopropane derivatives,, but their synthesis are not enantioselective since all derivatives are obtained in racemic forms. It is well worth noting that the cyclopropane-1,2-dicarboxylic acid occurs in three stereoisomeric configurations: The meso-cis and the (+)- and (-)- trans forms. The cis- acid whose the melting point is 139 °C (anhydride melting point is 59°C), is generated by decarboxylation of cyclopropane -1,1,2-tricarboxylic acid [7Conrad, M.; Guthzeit, M. On the action of α-β-dibromopropionic acid on malonic acid esters. Ber, 1884, 17, 1185.
[http://dx.doi.org/10.1002/cber.188401701314] ]. However, the reaction of methyl acrylate and methyl diazoacetate, after hydrolysis permits to obtain a mixture of cis and trans –cyclopropane -1,2-dicarboxylic acid whose the melting point is 175 °C, which fails to give an anhydride on treatment with acetyl chloride [8Buchner, E. Influence of Diazoessigäther on the Aether Unsaturated Acids. Ber, 1890, 23, 701.
[http://dx.doi.org/10.1002/cber.189002301113] ].
The trans-acid is obtained also from pyrolysis of the pyrazoline resulting from the addition of diazomethane to dimethyl fumarate [9Pechmann, H.V. Pechmann HV. Ueber Diazomethan. European Journal of Inorganic Chemistry. Ber., 1894, 1;27(2), 1888-91.].The cyclopropanedicarboxylic acids are inter-convertible under conditions where the cis –anhydride can be formed, it’s the case of the normal pyrolysis, where the trans is converted into the cis form. However, if anhydride formation is prevented as in the case of alkali fusion, the cis is converted into the more stable trans form [10About some polycarboxylic acids of trimethylene. Buchner, Ann., 1895, 284, 197.].
Decarboxylation of cyclopropanedicarboxylic acids has been the subject of several studies, and the data suitable to interest the forerunner researchers in the decarboxylation within acid medium have included the studies in protic medium in the presence of mineral acids, or the decarboxylation without solvent for which the carboxylic protons represent the acid medium. Whereas, the important data concerning the decarboxylation in basic medium have treated in particular the reactions in the presence of base and polar aprotic solvents.
According to the Abell and Lennon’s study [11Abell, P.I. Kinetics of the Decarboxylation of Some 1,1-Cycloalkanedicarboxylic Acids. J. Org. Chem., 1965, 30, 4212.
[http://dx.doi.org/10.1021/jo01023a052] , 12Abell, P.I. Stereochemistry of the Decarboxylation of Some 1,1,2-Cycloalkanetricarboxylic Acids. J. Org. Chem., 1965, 30, 1206.
[http://dx.doi.org/10.1021/jo01015a059] ], the mechanism of decarboxylation in protic acid medium, proposed for malonic acid [13Franckel, G. Decarboxylation of Malonic Acid in Quinoline and Related Media. J. Am. Chem. Soc., 1954, 76, 15.] Scheme. (1), was revealed difficult to be transposable to cyclopropanic derivatives.
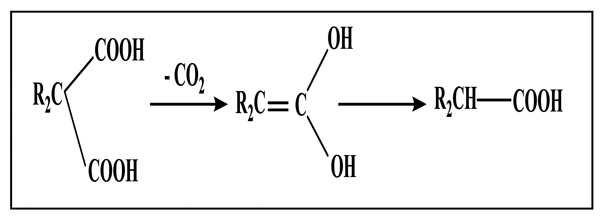 |
Scheme (1) Decarboxylation mechanism in protic acid medium. |
In fact, the generation of an exocyclic double bond would increase the reactivity of small cycle in order to make the transfer of invoked hydrogen for the cyclic intermediate d very easy towards the probable structure as it is shown in following scheme:
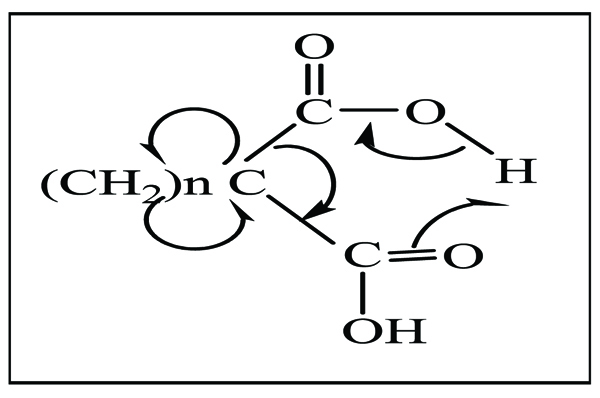 |
d Compound |
Pr. S. Benayachevia have reported that the most probable way, interpreting the different mechanisms involved in the reactions of cyclopropanic derivatives, is strongly based on the lactone formation during the decarboxylation without solvent, of several cyclopropanedicarboxylic-1,1 and tricarboxylic-1,1,2 acids. Therefore, the diacid b (Scheme. 2), was obtained from hydrolysis of the cyclic ester a, when the diacid loses the carbon dioxide by heating, and leads to formation of colored oil in which three products were separated: Stereoisomeric monoacid c in very low yield and the lactone d as the major product [14Addition Reactions of Vinyl Phenyl Ketone. III. Methyl Malonate. J. Am. Chem. Soc., 1933, 55, 2953.].
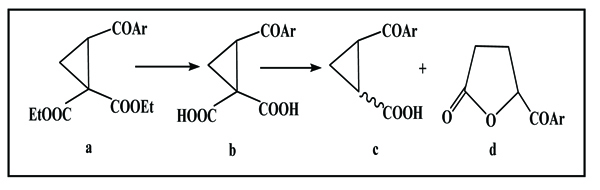 |
Scheme (2) Decarboxylation without solvent of cyclopropane-1, 1-dicarboxylic acids. |
Moreover, cyclopropanic-1,1,2 acid a’ (Scheme. 3) undergoes the cleavage of propanic ring that results in the formation of 3-oxabicyclo [3.1.0] hexane-2,4-dione and the corresponding lactone: paraconic acid c’, the latter releases a molecule of water and leads to the formation of citraconic anhydride d’.
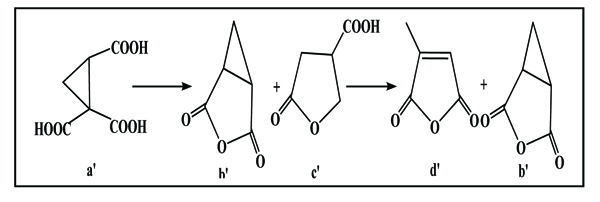 |
Scheme (3) The cleavage of propanic ring of cyclopropanic-1,1,2 acid. |
The kinetic investigation of J.Bus, H. Steinberg, and Th.T de Boer has rigorously interpreted the mechanism which is related to the decarboxylation and the opening of propanic ring . Otherwise, the decarboxylation of geminal dicarboxylic acids in diluted H2SO4, H2O and aqueous NaOH, generated the corresponding γ-butyrolactone in good yield [15de Boer, Th. J. The isomerisation and decarboxylation of cyclopropane-1,1-dicarboxylic acids. Tetrahedron letters, 1966, 7(18), 1905, 2048.].
2. RESULTS AND DISCUSSION
The preparation of new enantioselective 1,5-dimethyl - 3-oxabicyclo [3.1.0] hexane-2,4-dione (compound 5) was achieved from the action of 2-chloro propionic acid (compound 1) and ethyl methacrylate (compound 2) according to the following protocol (Scheme. 4).
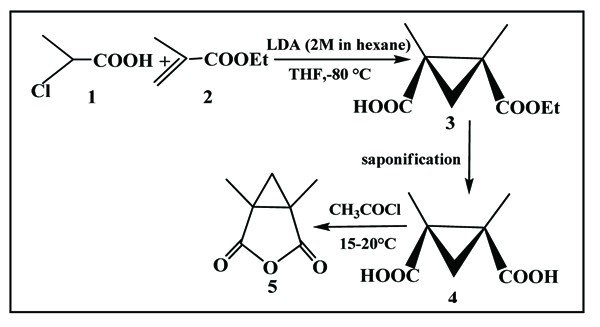 |
Scheme (4) Preparation of new enantioselective 1,5-dimethyl -3-oxabicyclo [3.1.0] hexane-2,4-dione. |
In fact, huge efforts were devoted to generate the compound 5 using many other methods in synthesis, described in the literature, so we have focused our work steps on some of which we consider the most convenient trends in our field:
2.1. Synthesis of Gerard Bonavent et al. Scheme. (5) [16Bonavent, G.; Zoller, M.C.; Guitard, M.; Jullien, R.F. Synthesis of polyfunctional cyclopropane derivatives. Bull. Soc. Chim. Fr., 1964, 10, 2462-2471.]
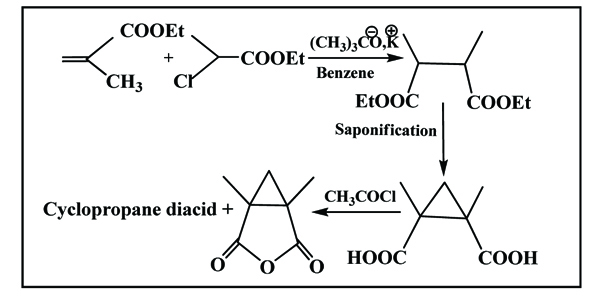 |
Scheme (5) Gerard Bonavent’s method. |
2.2. From Phosphore Oxychloride Scheme. (6) [17Von der Saal, W. Cyclopropanediamines. 3. Pure diastereomers of 1,2-cyclopropanedicarboxylic acids and derivatives. Liebigs Ann. Chem., 1989, 8, 703-712.
[http://dx.doi.org/10.1002/jlac.198919890218] ]
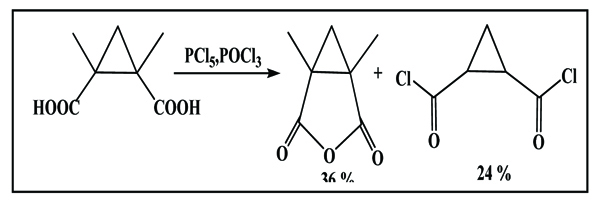 |
Scheme (6) Synthesis using phosphore oxychloride reagent. |
2.3. From Diazomethane Scheme. (7) [18Carl, R. Alkylation, acylation, and cyclopropanation reactions of α-halo carboxylic acid dianions. Synthesis, 1982, 4, 284-286.]
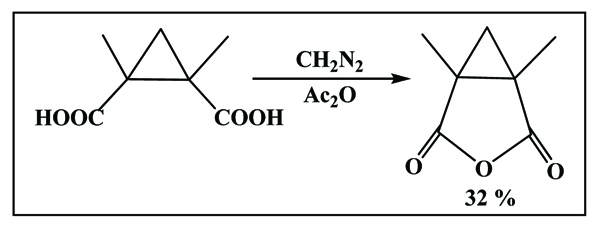 |
Scheme (7) Synthesis using diazomethane reagent. |
2.4. New Access Method in the Synthesis of 1,5-Dimethyl -3-Oxabicyclo [3.1.0] Hexane-2,4-Dione
The proposed synthesis pathway has provided the access to expected cyclopropanic anhydride, by condensation of 2-chloropropanoic acid with ethyl methacrylate, (principle of Michael reaction) and formation of carbanion that is able to attack the double bond, activated by electrophilic group, according to the following protocol (Scheme. 8)
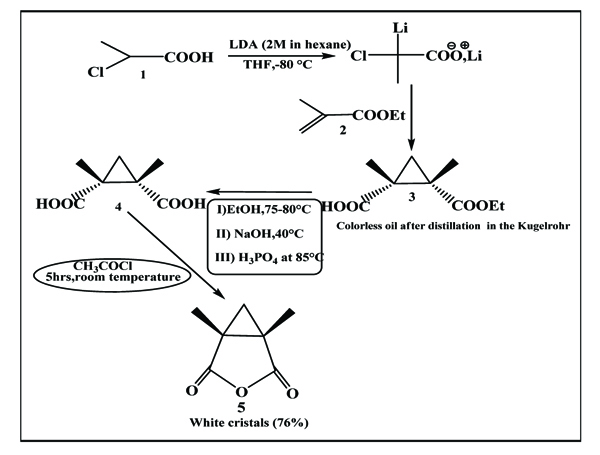 |
Scheme (8) Condensation of 2-chloropropanoic acid with ethyl methacrylate. |
3. EXPERIMENTAL SECTION
3.1. General
The 1H NMR spectra were recorded on Brücker DPX 400.13 MHz for proton and 100.62 MHz for 13C NMR using TMS as internal standard. The IR spectra were recorded on Perkin Elmer FT-IR spectrometer SPECTRUM 1000, on samples packaged in pellets of KBr or in the form of a deposit between two pellets of KBr (25 × 4 nm). Wavelengths were measured in cm-1.
3.2. Materials
All compounds, 2-chloropropanoic acid, ethyl methacrylate, THF, LDA, CH3COCl, Hexane and EtOH as well as inorganic compounds are commercial products and used without further purification.
All reactions and handling were carried out in a well-ventilated hood. Progress of the reactions and purity of the compounds were controlled by thin layer chromatography on silica gel (60 F254 Merck and 0.2 mm thick). The compounds were mainly revealed using U.V apparatus as visualizing agent, then by soaking in a KMnO4 or I2 aqueous solution, followed by heating on a hot plate. The illuminated chromatographies were carried out with silica gel (40-63 µm). Evaporation of solvents was performed at reduced pressure, using Buchi rotary evaporator.
3.3. General Synthetic Procedure
3.3.1. Cis-1,3-dimethyl-1,2-Cyclopropanedicarboxylic Acid Monoethyl Ester
To a solution containing 66 mL LDA (2M) and THF/n-heptane at -80 °C, placed carefully a solution of 6.42 g (59 mmol, 5.43 mL) 2-chloropropanoic acid dissolved in 24 mL THF anhydride. After stirring for 30 minutes at the same temperature, 7.50 mL (6.88 g, 59 mmol, 1 eq) ethyl methacrylate was added. The mixture was left in stirring for 20 minutes at -80 °C, and was slowly heated till ambient temperature. 150 mL of aqueous citric acid (10%) was added. The mixture was extracted with ethyl acetate. The organic phase was dried by Na2SO4 and concentrated under vacuum to a dark brown oil which was subjected to Kügelrohr distillation (at 75°C, under vacuum of Edwards pomp), and obtaining an uncolored oil: 6.56 g The yield: 58.7%.
3.3.2. Cis-1,3-Dimethyl-1,2-Cyclopropanedicarboxylic Acid
To a solution containing 190 mg (1mmol) ethyl methacrylate in 0.25 mL ethanol (95%), heated at 80 °C to 85 °C, placed a solution of 0.25 mL (3.5 eq) NaOH (40%, 14.3M) dropwise. At the end of the addition, the mixture became a white flaky matter to which added 0.25 mL water and 1 mL ethanol (95%). The reaction was stirred at refluxing temperature for 12 hours and concentrated under vacuum. To the residue containing diacid and sodium phosphate, added the ethyl acetate and the reaction was stirred at refluxing temperature for a few hours. After cooling, the product was filtrated on celite paper three times, and concentrated under vacuum to light brown oil which is cyclopropanic diacid .
3.3.3 Cis-1,5-Dimethyl -3-Oxabicyclo [3.1.0] Hexane-2,4-Dione
To the 1,3-dimethyl-1,2-cyclopropanedicarboxylic acid (219 mg,1.4 mmol), added (330 mg,4.2 mmol, 0.3 mL) acetyl chloride . The reaction was left in stirring at 15-20 °C for 15 hours. The mixture was concentrated under vacuum and heated in Kügelrohr at 80°C -100°C (real temperature at the ball of apparatus. On the other hand, the one which was displayed at the dial (165 -180°C), related to the temperature at the bottom of apparatus) under vacuum of Edwards pomp. White crystals appeared at the first ball (163 mg) The yield: 54%.
3.3.4 Cis-1,5-Dimethyl -3-Oxabicyclo [3.1.0] Hexane-2,4-Dione
1H NMR (400 MHz, CDCl3): δ 1.81 (dd, 1H, J= 4.9,2.0 Hz, or d, J =4.9 Hz,CH2), δ 1.490 (s, 3H, Mc), δ 1.486 (s, or d, J = 2.0 Hz,3H,Mc), δ 1.22 (d, 1H, J = 4.9Hz, CH2)
13C NMR (100 MHz, CDCl3): δ 171.19(CO), δ 31.55 (C quart), δ 29.70 (CH2), δ 10.00 (CH3) .
IR, υ (KBr disc): 3093, 2991, 2985, 2977, 2943, 1861 cm−1.
CONCLUSION
Cyclopropane-1,2-dicarboxylic anhydrides present an efficient class of cyclopropane compounds which have found many applications in organic syntheses and afford certain values in industrial production.
Electronic and steric properties of cyclopropane derivatives, especially their conformational rigidity, which makes it possible to orient the functional groups in a perfectly defined system, generating a particularly important and interesting structural motif in medicinal chemistry.
The study of cylopropanic system was mainly carried out by remarkable achievement of the related results, underlining the reactivity of the cyclopropanic-1,2 anhydride in the presence of acetyl chloride at ambient temperature and thus the use of LDA during the condensation of 2-chloropropanoic acid with ethyl methacrylate gave rise to the synthesis and made it possible to describe the 1,5-dimethyl-3-oxabicyclo[3.1.0] hexane-2,4-dione, functionalized as cis-structure.
In conclusion, the simplicity of experimental procedure, the high efficiency of the corresponding reactions, as well as the regioselectivity of synthesis are the advantages of the reported protocol.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGMENTS
We gratefully acknowledge the University of Rennes for scientific training and the University of Mentouri-Brothers-Algeria for financial support.
REFERENCES
| [1] | Salaün, J.; Baird, M.S. Curr. Med. Chem., 1995, 2, 511. |
| [2] | Liu, H.W.; Walsh, C.T. Biochemistry of the Cyclopropyl Group, 1987, Chap 16; Kennewell PDbMatharu, S.S. J. Chem.Soc .Perkin Trans, 1982, 1, 2553. |
| [3] | a) M.Eliott . Synthetic pyrethroids ,ACS .Symposium Series ,No.42: Washington DC,1977. b) M.Eliott.; N.F.James .Chem.Soc.Rev, 1978, 7,473 . c) Imperial Chemical Industries Ltd.Brit, 1,537,449; Chem Abstr, 1979,91,51102 w. |
| [4] | a) McCoy, L.L. J. Am. Chem. Soc., 1958, 80, 6568. b) McCoy, L.L. J. Am. Chem. Soc., 1962, 84, 2246. c) McCoy, L.L. J. Org. Chem., 1960, 25, 2078.Fraisse, R. Bull; Soc.Chem: France, 1959, p. 1102. |
| [5] | a) Inouye, Y.; Horiike., Ms; Inouye, Y.; Inouye, Y.; Inouye, Y. J. Am. Chem. Soc., 1961, 83, 2962. b) Inouye, Y. Tetrahedron, 1968, 24, 2907. [http://dx.doi.org/10.1021/ja01474a048] [http://dx.doi.org/10.1016/S0040-4020(01)98699-5] |
| [6] | Schmidt, U. R.Schröer and A.Hochrainer. Liebigs Ann., 1970, 773, 180. [http://dx.doi.org/10.1002/jlac.19707330120] |
| [7] | Conrad, M.; Guthzeit, M. On the action of α-β-dibromopropionic acid on malonic acid esters. Ber, 1884, 17, 1185. [http://dx.doi.org/10.1002/cber.188401701314] |
| [8] | Buchner, E. Influence of Diazoessigäther on the Aether Unsaturated Acids. Ber, 1890, 23, 701. [http://dx.doi.org/10.1002/cber.189002301113] |
| [9] | Pechmann, H.V. Pechmann HV. Ueber Diazomethan. European Journal of Inorganic Chemistry. Ber., 1894, 1;27(2), 1888-91. |
| [10] | About some polycarboxylic acids of trimethylene. Buchner, Ann., 1895, 284, 197. |
| [11] | Abell, P.I. Kinetics of the Decarboxylation of Some 1,1-Cycloalkanedicarboxylic Acids. J. Org. Chem., 1965, 30, 4212. [http://dx.doi.org/10.1021/jo01023a052] |
| [12] | Abell, P.I. Stereochemistry of the Decarboxylation of Some 1,1,2-Cycloalkanetricarboxylic Acids. J. Org. Chem., 1965, 30, 1206. [http://dx.doi.org/10.1021/jo01015a059] |
| [13] | Franckel, G. Decarboxylation of Malonic Acid in Quinoline and Related Media. J. Am. Chem. Soc., 1954, 76, 15. |
| [14] | Addition Reactions of Vinyl Phenyl Ketone. III. Methyl Malonate. J. Am. Chem. Soc., 1933, 55, 2953. |
| [15] | de Boer, Th. J. The isomerisation and decarboxylation of cyclopropane-1,1-dicarboxylic acids. Tetrahedron letters, 1966, 7(18), 1905, 2048. |
| [16] | Bonavent, G.; Zoller, M.C.; Guitard, M.; Jullien, R.F. Synthesis of polyfunctional cyclopropane derivatives. Bull. Soc. Chim. Fr., 1964, 10, 2462-2471. |
| [17] | Von der Saal, W. Cyclopropanediamines. 3. Pure diastereomers of 1,2-cyclopropanedicarboxylic acids and derivatives. Liebigs Ann. Chem., 1989, 8, 703-712. [http://dx.doi.org/10.1002/jlac.198919890218] |
| [18] | Carl, R. Alkylation, acylation, and cyclopropanation reactions of α-halo carboxylic acid dianions. Synthesis, 1982, 4, 284-286. |