- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Arthritis Journal
(Discontinued)
ISSN: 1876-5394 ― Volume 7, 2014
Macrophage Subsets in Immune-Mediated Inflammatory Disease: Lessons from Rheumatoid Arthritis, Spondyloarthritis, Osteoarthitis, Behçet’s Disease and Gout
Maria Cristina Lebre*, Paul Peter Tak
Abstract
Macrophages are a major cell population in most of the tissues, and their numbers increase massively in inflammation, in wound healing and in tumors. In particular, macrophages contribute to autoimmune events in rheumatic diseases, such as rheumatoid arthritis (RA) or spondyloarthritis (SpA), mainly acting as antigen-presenting cells and also as the major source of inflammatory mediators that are important in joint inflammation. In this respect, macrophages release a variety of pro-inflammatory cytokines and chemokines and events downstream of this cytokine cascade will contribute to cartilage and bone destruction. It is becoming clear that differential macrophage activation by distinct mechanisms is crucial for their function. This review will discuss several aspects of macrophage function in immune-mediated inflammatory disease with particular emphasis in RA, SpA, osteoarthitis, Behçet’s disease and gout.
Article Information
Identifiers and Pagination:
Year: 2010Volume: 3
First Page: 18
Last Page: 23
Publisher Id: TOARTHJ-3-18
DOI: 10.2174/1876539401003010018
Article History:
Received Date: 18/6/2009Revision Received Date: 2/8/2009
Acceptance Date: 3/8/2009
Electronic publication date: 12/1/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Academic Medical Center/University of Amsterdam, Division of Clinical Immunology and Rheumatology, K0-154, P.O. Box 22700, 1100 DE Amsterdam, The Netherlands; Tel: +31 20 5667688; Fax: +31 20 6919658; E-mail: c.lebre@amc.uva.nl
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 18-6-2009 |
Original Manuscript | Macrophage Subsets in Immune-Mediated Inflammatory Disease: Lessons from Rheumatoid Arthritis, Spondyloarthritis, Osteoarthitis, Behçet’s Disease and Gout | |
MACROPHAGES: INTRODUCTION
Macrophages are the first line of defense of the organism against pathogens and, in response to the microenvironment, become differentially activated. It has become increasingly clear that macrophages are not a homogenous population but one that can be divided into specific, although overlapping, subsets according to their polarization requirements, phenotype, and function [1, 2]. Classically activated macrophages (M1) are the main source of soluble pro-inflammatory cytokines, such as tumor necrosis factor-a (TNF-α) and interleukin-1β (IL-1β), whereas alternatively activated macrophages (M2) have been implicated in immune regulation, phagocytosis, and tissue remodeling (Fig. 1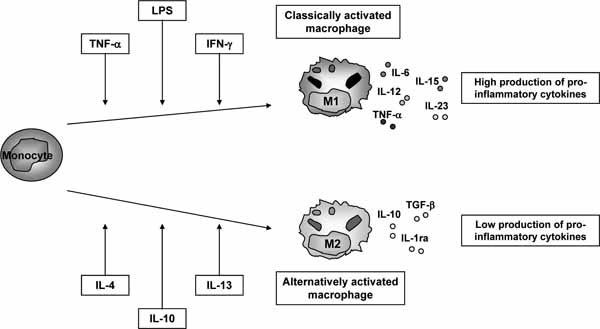 ). The regulation of macrophage populations, in particular their numbers in RA synovium during disease progression and following therapy has been recently reviewed [3].
). The regulation of macrophage populations, in particular their numbers in RA synovium during disease progression and following therapy has been recently reviewed [3].

MACROPHAGES IN RA
RA is a common autoimmune chronic inflammatory joint disease, characterized by macrophage, plasma cell and lymphocyte infiltration into the synovium, accumulation of synovial fibroblasts, and joint destruction [4]. Discoveries of pro-inflammatory mediators such as TNF-α and other secretory products have provided valuable insights into the role of macrophages in many acute and chronic disease processes, leading to the development of effective therapeutics.
Macrophages are critically involved in the pathogenesis of RA. Macrophages not only produce a variety of pro-inflammatory cytokines and chemokines in response to pathogens and cytokines, but they also contribute to the cartilage and bone destruction in RA through multiple mechanisms [5]. In normal synovium, the synovial lining layer is comprised of a cell layer of maximal 1-3 layers of cells. Within this synovial lining two cell types can be found: intimal macrophages and fibroblast-like synovial cells [6]. Specifically, macrophages are important mediators of chronic inflammation and are prominent in the synovial lining and sublining of patients with RA. In RA synovium, the lining layer becomes thickened due to increased numbers of macrophages and lymphocytes, and proliferated fibroblast-like synoviocytes [4, 7, 8]. Of interest, the intimal macrophages are more highly activated than sublining macrophages and express higher amounts of TNF-α and IL-1α (M1 characteristics). They are thought to migrate into the expanding pannus (hanging flap of tissue that forms in the joint affected by the disease, causing loss of bone and cartilage) that participates in the degradation of articular cartilage and subchondral bone [9].
CD68 is a 110-kD transmembrane glycoprotein that is highly expressed by human monocytes and tissue macrophages. This marker is widely used to identify macrophages both in the blood and tissue. Of importance, changes (e.g. decrease) in numbers of CD68 positive synovial sublining macrophages correlate with clinical improvement independently of the therapeutic strategy [10, 11]. It has been demonstrated that the change in the numbers of sublining macrophages can be used to predict possible efficacy of new anti-rheumatic treatments for RA [11, 12] whereas the number of CD3 positive T cells appears to be a better marker to predict possible efficacy of new anti-rheumatic treatments for psoriatic arthritis (PsA) [13].
Toll-like receptors (TLRs) present on macrophages and dendritic cells (DC) recognise pathogen-derived products and lead to the induction of the production of effector molecules that regulate innate and adaptive immune responses [14, 15]. The potential role of TLRs in RA pathogenesis has been supported by the demonstration of TLR2, TLR3 and TLR4 expression in RA synovial tissue [16-20]. The expression of TLR2 and TLR4 was increased on CD14+ macrophages from the joints of RA patients compared with that on control in vitro-differentiated macrophages or control peripheral blood monocytes [21]. Neither TLR2 expression nor TLR4 expression differed between RA and other forms of inflammatory arthritis. However, TNF-α and IL-8 expression was increased on RA synovial fluid macrophages that were stimulated in vitro with peptidoglycan (TLR2 ligand) or LPS (TLR4 ligand) compared to synovial fluid macrophages obtained from patients with other forms of inflammatory arthritis or with control macrophages [21]. In a different study, it was demonstrated that RA blood-derived macrophages have increased TLR2 and TLR4 expression and increased response to microbial TLR2 and TLR4 [22]. In this respect, the presence in RA synovium of peptidoglycan-positive antigen-presenting cells (APCs), that include synovial macrophages and mature DC, is related to cytokine production in vivo [23]. In accordance synovial APC-derived IL-12 and IL-18 (M1 mediators) is associated with synovial TLR2 and TLR4 expression [17]. In addition, to the potential role of TLR ligands, a number of endogenous stress response proteins have been implicated as potential TLR ligands in RA. In this respect, the 96-kDa heat shock glycol-protein (gp96) has been reported to activate macrophages [24-27] and DC [28-30] promoting the release of the pro-inflammatory cytokines TNF-α and IL-1β [31] (M1 mediators). Interestingly, the expression of this protein is increased in RA synovial tissue and fluid compared to synovium from osteoarthritis (OA) and arthritis-free controls [22]. Moreover, the expression of TLR2, but not TLR4, on synovial fluid macrophages was strongly positive correlated with the level of gp96 in the synovial fluid suggesting a potential role for gp96 as an endogenous TLR2 ligand in RA. Altogether these data suggest that activated APCs may contribute to inflammation within the microenvironment of the joint in RA patients. Moreover, since both endogenous and exogenous TLR ligands are thought to contribute to the chronic inflammation observed in RA the design of new strategies that disrupt the interaction between TLR and their ligands might be of potential importance in order to diminish the inflammatory process.
To date, there is no known marker that specifically distinguishes tissue macrophages present in normal synovium (that play a role in homeostasis by removing for instance dead cells) and macrophages present in inflammed synovium (that produce exaggerated amounts of mediators involved in inflammation). Of interest, the Z39Ig protein (complement receptor for C3b and iC3b) is expressed on resident tissue macrophages in various tissues. The expression of the Z39Ig protein was limited to intimal macrophages in normal, RA, OA and PsA synovium. The numbers of Z39Ig+CD11c+ cells and the ratios of Z39Ig+CD11c+ cells to Z39Ig+ cells were increased in the intimal lining layer of RA as compared with those of OA and PsA [32]. Moreover, in vitro studies showed that expansion of Z39Ig+CD11c+ cells was characteristic of RA intimal lining layer and suggests that alternation of macrophage differentiation during the late stages of disease may be present in RA synovitis (i.e. the differential expression of this protein by macrophages at more advanced maturation status). These findings also support the hypothesis that the increased numbers of Z39Ig+CD11c+ cells are valuable for the identification of RA in early arthritis [32].
Altogether, it is clear that the characterization of molecules expressed by macrophages that regulate and/or phenotypically define their functions in RA pathogenesis is of crucial interest. Furthermore, achieving a detailed understanding of macrophage function(s) in RA holds potential for modulating macrophage expressing markers and/or soluble mediators in order to down-regulating the autoimmune response.
MACROPHAGES IN SPONDYLOARTHRITIS (SpA)
Spondyloarthritides (SpA) is the overall name for a family of inflammatory rheumatic diseases that comprises ankylosing spondylitis (AS), psoriatic arthritis (PsA), reactive arthritis, enteropathic-related arthritis and undifferentiated SpA. Specifically, the axial disease affects the spine, the sacroiliac joints and the hips while the peripheral disease includes arthritis, with a preference for asymmetrical inflammation of joints of the lower limbs and enthesitis [33]. Additionally, SpA is often characterized by subclinical inflammation of the gut which partially resembles inflammatory bowel disease. In general, SpA patients are negative for rheumatoid factor hence the term ‘seronegative’, and collectively, SpA shows an association with carriage of the HLA-B27 gene. Enthesitis, which is inflammation of the attachment of tendons, ligaments and joint capsules to bone, is the hallmark of SpA [34, 35].
PsA is a chronic, progressive disease in most patients [36]. A polyarticular onset of PsA is associated with a more destructive course [37]. The synovial infiltrate of patients with PsA and RA is comparable with regard to numbers of fibroblast-like synoviocytes and macrophages [38]. Prominent swelling and inflammation of peripheral joints is a hallmark of both RA and SpA, with a preference for large joints of the lower extremities in SpA. Of interest, the total number of macrophages is similar in RA and SpA synovitis, but the subset expressing the M2 surface marker, CD163 [39, 40], is clearly increased in the latter [41-43]. In accordance with this observation, it was recently described that synovial fluid from SpA patients promotes preferential expression of the M2 markers CD163 and CD200R in vitro. Interestingly, this was still observed even if synovial fluid levels of the prototypical M2-polarizing factors (IL-4, IL-13, and IL-10) were not increased as compared with those in RA synovial fluid [44].
Once again it is becoming clear that the composition of the inflammatory milieu, which is characteristic of a particular disease, will determine the existence and the generation of a defined macrophage population with likely specific functions. In this respect RA macrophages are associated with a M1 phenotype whereas SpA macrophages with a M2 phenotype.
MACROPHAGES IN OSTEOARTHRITIS (OA)
OA is characterized by loss of articular cartilage and modification of subchondral bone. Pain in and impaired movement of joints affected by OA cause disability. Fluid accumulation (joint effusion), bone overgrowth (osteophytes), and weakness of tendons and muscles can also result from the degenerative process. OA commonly affects the hands, feet, spine, and large weight-bearing joints, such as the hips and knees. The pathogenesis of OA is largely unknown and under current investigation. Biomechanical stress (especially static compression) appears to be an important factor in the pathogenesis of OA, but it cannot completely explain the disease process [24]. Moreover, it is now believed that cartilage integrity is maintained by a balance obtained from cytokine-driven anabolic and catabolic processes. In OA, the specific causative for the pathological process has not been identified, but synovial inflammation at the clinical stage is now well-documented phenomenon [45].
Morphological changes observed in OA include cartilage erosion as well as a variable degree of synovial inflammation. Current research attributes these changes to a complex network of biochemical factors, including proteolytic enzymes that lead to a breakdown of the cartilage macromolecules. Cytokines such as IL-1β and TNF-α produced by activated synoviocytes, mononuclear cells (e.g. macrophages) or by articular cartilage itself significantly up-regulate metalloproteinases (MMP) gene expression [45, 46].
In experimental OA the involvement of macrophages in this pathology has been demonstrated [47, 48]. In a study in which synovial lining macrophages in the knee joint were selectively depleted prior to elicitation of collagenase-induced experimental OA, it was observed that macrophages mediate osteophyte formation and fibrosis in the early stages of experimental OA [49]. Moreover, these synovial macrophages were crucial in early MMP activity and appeared to mediate MMP production in synovium rather than cartilage [50] reinforcing the importance of these cells in OA inflammation.
Recently the involvement of the Wnt signaling pathways (that are involved in embryonic development of cartilage and bone) in human and experimental OA has been demonstrated [51]. In particular, Wnt-induced signaling protein 1 (WISP-1) expression was strongly increased in the synovium and cartilage of human and experimental OA. These findings suggest that WISP-1 expression might be a feature of experimental and human OA and demonstrated the regulation of chondrocyte and macrophage MMP and aggrecanase expression by WISP-1 and that this molecule was capable of inducing articular cartilage damage in models of OA [51].
In OA macrophage derived-MMP and pro-inflammatory cytokines might contribute to cartilage destruction and are likely to play a crucial role in disease pathogenesis. But are OA macrophages more associated with a M1 phenotype?
MACROPHAGES IN OTHER FORMS OF INFLAM-MATORY ARTHRITIS
Behçet Disease (BD)
Behçet disease (BD) is a chronic, relapsing, inflammatory disorder which is clinically characterized by bipolar aphtosis and microvascular skin and ocular lesions [52]. Although several immunological abnormalities have been demonstrated, the exact mechanism of the inflammatory changes occuring are yet to be elucidated.
The synovitis of patients with Behçet in terms of CD68+ macrophage numbers is similar in the intimal lining layer as well as the synovial sublining to the synovitis of PsA patients [53]. However, it has been reported that serum factor(s) are able to induce classical (pro-inflammatory) activation (M1 phenotype) of human peripheral blood macrophages in vitro in this disease [54] suggesting that serum factor(s) might be responsible for inflammatory changes in BD.
Gout Arthritis
Gout is a disease that results from an overload of uric acid in the body. This overload of uric acid leads to the formation of tiny crystals of urate that deposit in tissues of the body, especially the joints. When crystals form in the joints it causes recurring attacks of joint inflammation (arthritis). Understanding how uric acid crystals provoke inflammation is crucial to improving the management of acute gout. It is well known that urate crystals stimulate monocytes and macrophages to produce inflammatory cytokines, but the tissue response of the synovium is less well understood. Recent studies suggest that monocyte/macrophage-derived IL-1b may also play a central role in gout [55, 56]. In this respect, monocytes or macrophages within the joint release IL-1b, which induces other cells within the joint, such as endothelial cells and fibroblasts, to produce cytokines and chemotactic factors, which result in the recruitment of neutrophils to the joint [57].
Moreover, pathogenic crystals induce the expression of a number of cytokines and chemokines from monocytes and macrophages. Of these, and as stated above IL-1 plays a pivotal role. Specifically, monosodium urate monohydrate (MSU) crystals promote the processing and release of IL-1β, and signaling by IL-1R which is critical for the development of MSU crystal-induced inflammation in vivo [57]. In addition, it was recently described in the same model that resident macrophages that have a highly differentiated phenotype, rather than infiltrating monocytes or neutrophils (contrary to the current dogma), are important for initiating and driving the early pro-inflammatory phase of acute gout characterized by IL-1α and IL-6 [58]. In a different study using the same model, the role of TLR2 and TLR4 present in bone-marrow-derived macrophages (BMDMs) has been addressed [59]. In this respect, BMDMs from TLR2-, TLR4-, and MyD88-deficient mice showed impaired uptake of MSU crystals and suppressed production of IL-1β, TNF-α, keratinocyte-derived cytokine/growth-related oncogene alpha, and transforming growth factor β1 in vitro. These observations implicate that murine host required TLR2, TLR4, and MyD88 for macrophage activation in response to MSU crystals. Moreover, these findings implicate innate immune cellular recognition of naked MSU crystals by specific TLRs as a major factor in determining the inflammatory potential of MSU crystal deposits and the course of gouty arthritis [59]. The molecular processes underlying the inflammatory conditions of gout were recently described [56]. Macrophages from mice deficient in various components of the inflammasome such as caspase-1, ASC and NALP3 are defective in crystal-induced (e.g. MSU) IL-1β [56].
It is clear that macrophage-derived IL-1β (M1-associated phenotype in gout?) is a pivotal mediator of acute gout and could become a potential therapeutic target.
CONCLUDING REMARKS
The plasticity of the macrophage phenotype is well know, and macrophage switching from a “pro-inflammatory” to an “anti-inflammatory”phenotype over time remains a viable mechanism of action for resolution of acute inflammation. It is assumed that the presence of a particular macrophage population (with a specific function) is dependent on the type of disease and/or clinical stage. However, these specific observations have not yet been reported. Therefore, it remains to be established the precise mechanism(s) and factor(s) involved in the shift between M1 and M2 in these diseases and explore the mechanisms by which a particular macrophage poplulation uses to resolve inflammation. These efforts in investigating the different macrophage populations present in several rheumatic diseases are of crucial interest in terms of therapy design since it will help to define specific and safe target therapies.
ACKNOWLEDGEMENTS
The authors wish to acknowledge Fundação para a Ciência e Tecnologia (SFRH/BPD/38899/2007 to M.C.L.) and also the Dutch Arthritis Foundation (2008, nr. 0701014, to P.P.T. and M.C.L.) for financial support.