- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Arthritis Journal
(Discontinued)
ISSN: 1876-5394 ― Volume 7, 2014
Dendritic Cell Biology in Animal Models of Arthritis
Robert Benson*, James Brewer, Paul Garside , Iain B. McInnes
Abstract
In recent years the treatment of rheumatoid arthritis (RA) has changed considerably with the advent of novel biologic agents that target cytokines such as TNF. The impact on clinical practice has been considerable with achievement of high hurdle endpoints and reduced articular damage. Unfortunately, remission that is long lasting is rarely achieved and almost never reached in the absence of chronic drug therapy. Thus, interest in what should be the critical objective of autoimmune therapeutics – the re-establishment of self-tolerance – has become increasingly prominent. Models of experimental arthritis have only just begun to reveal the intricacies of dendritic cell (DC) biology in RA. And while manipulation of antigen presenting cells such as DCs can be used in the suppression of experimental arthritis, the basic functions and mechanisms regarding their impact is mostly obtained indirectly by inference from other autoimmune and infectious studies. Indeed, our understanding of the contribution of DC biology to induction and perpetuation of RA is relatively ill defined. Here we discuss recent advances in understanding basic DC biology, their roles in, and impact on, experimental arthropathy and resulting therapeutic implications. It is essential that more research into the direct contribution of DC activity in RA is forthcoming, particularly as they could hold the key to development of antigen specific therapeutics. The major contributing factor to this knowledge deficit is the difficulty inherent in investigating what are most likely pre-clinical immunological events, an area more suited to study in animal models.
Article Information
Identifiers and Pagination:
Year: 2010Volume: 3
First Page: 24
Last Page: 31
Publisher Id: TOARTHJ-3-24
DOI: 10.2174/1876539401003010024
Article History:
Received Date: 15/5/2009Revision Received Date: 16/10/2009
Acceptance Date: 22/10/2009
Electronic publication date: 12/1/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/ which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Glasgow Biomedical Research Centre, Division of Immunology, Infection and Inflammation, Faculty of Medicine, University of Glasgow, 120 University Place, Glasgow G12 8QQ, Scotland, UK; Tel: 44 141 3308412; Fax: 44 141 337 3217; E-mail: benson@clinmed.gla.ac.uk
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 15-5-2009 |
Original Manuscript | Dendritic Cell Biology in Animal Models of Arthritis | |
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic, disabling, inflammatory autoimmune disorder in which immune mediated pathology not only drives joint destruction but promotes damage in extra-articular tissues such as the skin, heart and lungs, with attendant co-morbidity and accelerated mortality [1, 2]. The processes underlying initiation and perpetuation of RA are currently unclear: cytokines such as TNF are recognised to play a central role in pathogenesis implicating innate immune pathways and inflammation in effector biology [3]. However cell targeting therapeutics such as rituximab and abatacept that modulate primarily adaptive immunity are also efficacious in the clinic suggesting that antigen-specific autoimmunity may also be important. This expansion of novel therapeutics has been achieved in part through application of sophisticated molecular and cellular biology, greater availability of clinical tissue samples but importantly, the availability of animal models facilitating in vivo studies. The relevance of increasing our understanding of the basic biological systems underlying the pathophysiology of RA is directly borne out in the plethora of novel biologic and small molecule agents that are in development (e.g. those targeting janus kinases, syk kinases, IL-1, IL-6, GM-CSF, IL-17, IL-23p40, CD22). Despite this remarkable progress of recent years, not all patients respond adequately, with a substantial proportion remaining refractory to intervention. This fact, combined with the risks associated with general immunosupression, has ignited interest in what should be the critical objective of autoimmune therapeutics – the re-establishment of self-tolerance. Translated into clinical practice, this could translate to drug free remission, a phenomenon rarely achieved in clinical practice even with optimised current regimens.
The long recognised association of RA with the shared epitope contained in class II MHC molecules, together with the outcome of several recent genome wide association studies have highlighted the importance of antigen presentation to CD4+ T cells in RA. Thus PTPN22, CTLA4, CD40L, HLADR4 lie adjacent to disease associated genomic markers and are highly likely to associate with disease risk and perpetuation. These moieties are associated with optimisation or regulation of antigen presentation. Therefore, identification of the antigen-presenting cell (APC) responsible for priming auto-reactive T cells is of paramount importance. Armed with such knowledge, intervention strategies could be developed to subvert APC interactions with T cells and bring closer the realisation of resetting self-tolerance.
Dendritic cells (DCs) are considered the most efficient stimulator of naïve T cells, capable of priming strong responses to foreign antigens yet presenting self to maintain/induce tolerance. In this respect they are ideally situated to drive activation of auto-reactive T cells. Indeed it has been hypothesized that DCs presenting self-peptides are responsible for breach of self tolerance in RA [4, 5] and there is an increasing body of circumstantial evidence in support of this, principally the definition of activated DC populations in patient synovial tissue and fluid [6]. This remains to be demonstrated directly, as has been achieved in some other autoimmune diseases [7, 8], leaving our understanding of the contribution of DC biology to induction and perpetuation of RA relatively ill defined. The major contributing factor to this knowledge deficit is the difficulty inherent in investigating what are most likely pre-clinical immunological events, an area more suited to study in animal models. By corollary, such modeling would allow the therapeutic potential of these cells to be ascertained.
DCs AS POTENTIATORS OF AUTOREACTIVITY
DCs in murine models constitute a highly heterologous population of cells, but can be broadly defined by their expression of CD11c and ability to up-regulate MHC class II to support CD4+ T cell responses. Classically, the life cycle of a DC follows the migration of progenitor/immature DC from the bone marrow to tissue, tissue conditioning while sampling local antigens, followed by maturation and migration to the draining lymph node. The microenvironment where antigen is sampled dictates the resulting context in which it is presented to CD4+ T cells, namely pro-inflammatory or tolerogenic. Under normal/non-inflammatory conditions, steady state migration of tissue DCs to lymph nodes is believed to facilitate maintenance of self-tolerance through presentation of self-antigens. However, DC detection of innate danger signals derived by recognition of pathogenic agents or stress/physical damage to cells induces up-regulation of a host of costimulatory molecules required for efficient priming of CD4+ T cell responses – the consequence is effector immunity (Fig. 1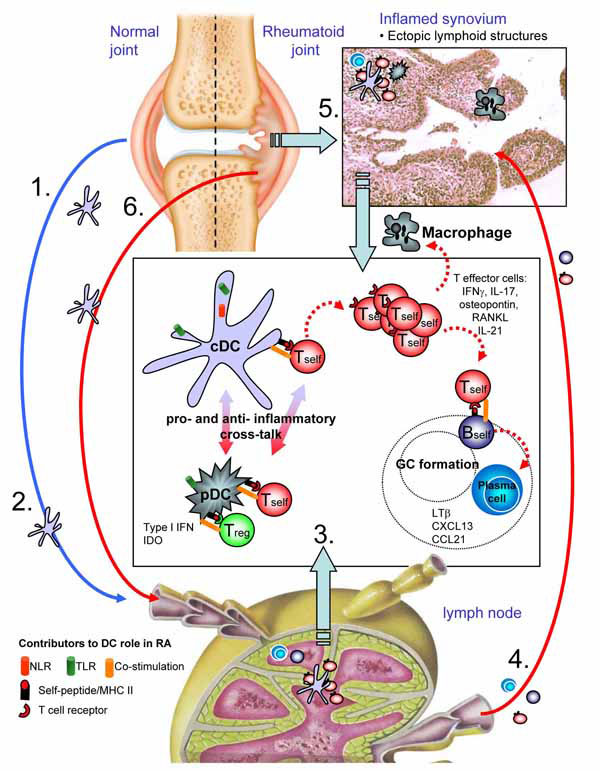 ).
).

Given this ability to dictate tolerance versus priming, DCs are ideally situated to drive aberrant activation of autoreactive CD4+ T cells that have escaped thymic selection, a process in which DCs also play an important role [9-11]. Thomas and Lipsky originally hypothesized that presentation of self-antigen by DCs could induce and maintain inflammatory responses in RA [4, 5], yet a definitive demonstration of this is still lacking. The first questions provoked by this hypothesis concern the physical nature or origin of the initiating antigen presented, when such presentation occurs and whether the same inciting antigen(s) drive perpetuation of disease? Secondly, given the heterogeneity of dendritic cell populations, which DC is central to these processes? Thirdly, what happens to the DC to induce presentation of self-antigen in a pro-inflammatory context? These questions are very difficult to address from the clinical perspective, especially given the likelihood that many of the events that require study occur prior to clinical presentation, potentially years before clear disease manifestation. In this regard, animal models offer a key advantage wherein the events preceding and maintaining autoimmune inflammation can be delineated and manipulated.
Antigens
Concerning the first question, the nature of the auto-antigen presented by DCs in RA - many models utilize immunization protocols to induce disease by using antigens identified in RA patients. In this circumstance the inciting antigen specificity is clear, yet what can be learned from this approach regarding which, and how, normally tolerated proteins become targeted auto-antigens is limited. What is clear from models where spontaneous autoreactive responses occur and through adoptive transfer of autoreactive T cells is that disease relevant self-antigens are constitutively presented. CD4+ T cells from donor mice suffering from CIA are able to induce disease in naïve recipients following adoptive transfer, indicating that processed collagen peptides are presented under normal circumstances. Proteoglycan specific TcR Tg and ZAP-70-/- T cells are also able to induce disease in syngeneic recipients [12, 13], however induction of pathology is only seen in lymphocyte deficient hosts indicating regulation of pathogenic T cells by the normal repertoire, perhaps through regulatory T cell activity or even homeostatic competition [14]. In either case, the target autoantigens are presented constitutively without demand for prior inflammation for their release. As regards which antigen? Available evidence suggests not only highly joint-relevant antigens such as aggrecan [12, 15], but even ubiquitously expressed proteins like glucose-6-phosphate isomerase can drive TcR transgenic T cell mediated arthropathy [14].
DC Populations
As in humans, murine DCs consist of a heterogenous population of antigen presenting cells. Two clearly distinct populations are readily identifiable, namely plasmacytoid (pDCs) and conventional DCs (cDCs). Greater subdivision of the cDC compartment reveals tissue/functional specific phenotypes including myeloid DCs and CD8α+ DCs with all populations expressing CD11c to some degree. Comparison of mice constitutively depleted of CD11c+ cells demonstrates the importance of DCs to development of autoimmunity, in part due to altered thymic selection, but also in peripheral regulation [9, 16].
CD11c+B220+PDCA-1+MHC class IIlo pDCs are a major source of type I interferon, vital in viral immunity. Although normally associated with protective immunity, pDCs have been ascribed an effector role in innate pathology seen in SLE [17] and in psoriasis. pDC detection of viral nucleic acids via Toll like receptor 7 (TLR7) and TLR9 stimulates MyD88 dependent pro-inflammatory cytokine production, IFNα, IFNβ, IFNγ, IFNω secretion and elevated expression of costimulatory molecules. Tolerance of self-nucleic acids released by either necrosis or apoptosis is predominantly thought to occur though their exclusion from endosomal compartments where TLR7 and 9 are located. This is in part mediated by extracellular degradation. Deficiency in DNAse I results in SLE type pathology that interestingly initially presents as a polyarthropathy [18]. In addition to innate responses, pDCs are also able to process and present captured antigen to CD4+ T cells, demonstrated both in vitro and in vivo (reviewed in [19]). Such data suggest that pDCs could contribute to autoimmune pathology in RA, however other evidence is to the contrary. Monoclonal antibody mediated depletion of pDCs in a preclinical model of RA revealed that these cells are not responsible for inciting autoimmune responses [20]. In this model, a Th1 response mounted to the irrelevant antigen ovalbumin (OVA) gave rise to anti-type II collagen specific T and B cell responses, with anti-CCP-antibodies and rheumatoid factor (RF) also detectable in serum. Depletion of pDCs prior to and during stimulation of the Th1 response had no bearing on the OVA recall response but resulted in exacerbation of the aforementioned autoimmune responses. Whereas no direct mechanism was identified for this regulation, pDCs were able to inhibit effector T cell cytokine production in an antigen dependent manner. Similar findings have been demonstrated in EAE [21], suggesting a delicate interplay between T cell priming by cDCs and regulation by pDCs, yet the molecular mechanism remains elusive. Human studies demonstrate that synovial pDCs express high levels of indoleamine 2,3-dioxygenase (IDO) [22], an enzyme that degrades tryptophan and can promote immunological tolerance. Inhibition of IDO can breach self-tolerance and promote rejection of tumours and grafted tissues [23-25]. However, use of the competitive inhibitor 1-methyl-tryptophan (1-MT) did not reverse regulation of EAE by pDCs [21], with the authors suggesting a balance between pDC regulation and cDC stimulation of autoreactive T cells, also suggested by the aforementioned OVA induced arthritis model [20].
Studies that directly address the contributing role of cDCs to development of experimental arthritis are few. Some evidence supporting the role of cDCs in driving experimental arthritis has arisen in the collagen induced arthritis model (CIA) [26]. Collagen pulsed bone marrow derived cDCs were sufficient to induce erosive arthritis when used to challenge susceptible DBA/1 mice [27]. Following joint-distal-injection, CII pulsed DCs could be detected in draining lymph nodes adjacent to proliferating T cells. Challenge in this way was sufficient to overcome endogenous regulatory mechanisms in the recipient mice, inducing CII specific T cell responses and production of CII-specific antibodies. Significant articular damage was also observed, with histological analysis revealing synovial infiltrate, hyperplasia and cartilage erosion. Transfer of cDCs in this manner, and the ensuing pathology, is consistent with findings in EAE [8]. Although DC-induced arthritis was not polyarticular, mice that had been primed previously in the contralateral footpad developed articular inflammation following rechallenge with unpulsed DCs or TNF alone. This might have been due to endothelial cell activation by the transferred DCs allowing access by primed collagen specific T cells. More recent investigation of cDC induction of CIA has further defined an in vivo population driving CIA in a similar manner, demonstrating that transfer of CD8α+ cDCs from arthritic mice drove disease in recipient DBA/1 mice [28]. This population of DCs is well known for its ability to cross-present antigen, however it has also been shown to present self-antigen [29-31], albeit in a tolerogenic context. Deregulation of this activity may further promote autoimmune reactivity.
Activation of DCs as Autoimmune Mediators
This leads us to question why DCs switch from a tolerogenic phenotype to prime autoreactive T cells. Epidemiological data have implicated prior infection with subsequent development of RA. In this context pattern recognition receptors (PRRs) are likely to be important in the activation of DCs potentially laden with self-antigens. In turn, pathogen activation of DCs laden with self-antigen does not routinely result in autoimmunity likely reflecting effective thymic education and peripheral regulatory mechanisms. Yet pathogen derived molecules are capable of inducing experimental arthritis. For example, stimulation of TLR2, 3, 4, 9 can initiate and/or and aggravate numerous models of arthritis, for example those driven by streptococcal cell wall [32], dsRNA, LPS [33, 34] and CpG DNA [35]. These studies clearly define roles for specific TLRs. However, it may be worth reconsidering the context in which we stimulate PRRS with excess of exogenous ligand - do such studies truly mimic how self-antigens may be presented leading to loss of tolerance?
Endogenous, in addition to microbial, ligands have been implicated in the pathogenesis of RA. Heat shock proteins (HSP60, 70) [36-38], the extracellular matrix component biglycan [39], heparin sulphate [40] and the extracellular domain of fibronectin [41] have all been shown to bind TLRs. Use of Bartonella Quintana LPS as an antagonist of TLR-4 signalling in ongoing CIA reduced clinical and histological signs of disease [42]. Inhibition had little effect on the anti-CII-antibody titres – however, this does not necessarily mean that there was no effect on the adaptive immune response. The aggressive immunization protocol employed in the CIA model produces high titres of anti-CII-antibodies; inhibition of TLR-4 in this system may not impinge on this already strong response. Examination of T cell responses to CII and perhaps induction of anti-CCP-antibodies in this context would be of interest. Mice deficient in RP105 (CD180), a TLR homologue lacking the Toll-IL-1 receptor domain, displayed exacerbated CIA pathology [43]. RP105 lacks the capacity to signal but in complex with MD-1 [44] can regulate TLR-2 and TLR-4 activity [45-47]. Normally CIA immunisation employs complete Freud’s adjuvant, the mycobacterial content of which is rich in TLR-2 and -4 ligands. Intriguingly, the authors demonstrated that RP105 deficient mice still developed a higher incidence and severity of CIA when incomplete Freund’s adjuvant was used, implicating the release of endogenous TLR ligands [43]. DCs from these mice were found to be more potent inducers of T cell IFNγ than wild type counterparts, highlighting the importance of this pathway to stimulating adaptive responses in autoimmune arthropathy.
While TLRs are important for recognition of extracellular and endosomal stimuli, Nod-like-receptors (NLRs) detect microbial products in the cytosol. The generation of NLRs, caspases and adaptor protein macromolecular structures/inflammasomes is important for example for pro-cytokine conversion to proinflammatory mediators (IL-1 and IL-18), cellular repair and survival (reviewed in [48]). Of note is the relation of NLRP3 inflammasome with recognition of uric acid released by necrotic cells, having implications in the pathology of gout-associated arthropathy [49]. As studies in murine systems reveal more about inflammasome biology and its ability to promote adaptive immunity [50], this aspect of DC recognition of pathogens may prove increasingly relevant to autoimmune arthritis.
The negative regulation of DC function appears equally important in preventing autoimmune arthritis as those pathways responsible for their stimulation. Mice deficient for DCIR (Dendritic Cell ImmunoReceptor) spontaneously developed autoimmunity through the excess DC expansion [51]. Expression of this c-type lectin containing a carbohydrate binding domain and an immunoreceptor tyrosine-based inhibitory motif (ITIM) is believed to negatively regulate DC function [51, 52]. Induction of CIA in DCIR deficient chimeric mice using a mild regime consisting of a single type II collagen/CFA immunization resulted in significantly increased disease severity compared to DCIR sufficient controls [51]. The potential importance of these findings to human disease is highlighted by the identification of a DCIR single nucleotide polymorphism associated with anti-CCP-negative RA [53, 54]. The contribution of the c-type lectin superfamily and similarly novel genes encoded in the antigen-presenting lectin-like receptor complex (APLEC) will no doubt reveal more about DC function in polyarthritic disease, indeed the aforementioned study drew a great deal of parallels between findings in oil induced arthritis in rats and human genomic studies [53].
The cytokine milieu resulting from inflammation will likely dictate how cDCs perpetuate chronic articular inflammation. The contribution of prostanoids such as PGE2 to RA have been of interest for many years, being found in high levels in synovial fluid and tissue of patients [55]. Prostanoids have been well characterized in CIA, with severity of disease being heavily impaired by deficiency in the EP2 and EP4 [56]. PGE2 signaling through these receptors was shown to switch bone marrow derived DC production of IL-12 to IL-23, driving IL-17 rather than IFNγ secretion by CD4+ T cells [57, 58]. Consistent with this, in vivo administration of a PGE1 analog agonist exacerbated CIA, elevating joint and lymph node levels of IL-17 and increasing serum titres of anti-CII-antibodies [58]. An additional compounding negative factor to prostanoid influence on DC biology in RA may lie in the ability of PGE2 to increase DC survival [59]. This is of particular relevance given that DC apoptosis aids maintenance of peripheral tolerance: in vivo overexpression of a baculovirus caspase inhibitor results in autoimmunity [60].
DC and Autoreactive T Cell Interactions
The stimulation/regulation of T cell responses by DCs employs a complex highly regulated series of adhesion and co-stimulatory molecules/cytokines in addition to the recognition of specific peptide/MHC class II. Many basic immunological mechanisms relevant to T cell/DC interaction have been identified but their relevance in the context of RA is only now becoming clear. The importance of peptide/MHC class II to development of experimental arthritis was elegantly demonstrated by blocking pathogenic T cell/APC interaction by using CII peptide/MHC class II multimers in CIA [61]. Development of CIA in HLA-DR1 transgenic mice was also disrupted when presentation of CII259-273 was inhibited. The authors demonstrated that CII accumulation of CII in the late endosomes of DCs and macrophages could be prevented by inhibiting macropinocytosis, preventing recognition by CII259-273/HLA-DR1 restricted hybridomas. This effect was reproduced in vivo, where administration of amiloride at disease onset prevented CII-specific-polyclonal T cell proliferative responses.
Given the complexity of identifying T cell epitopes, particularly in the context of RA heterogeneity in an ‘outbred’ human population, it is unsurprising that therapeutic strategies are focused on other conserved pathways involved in DC/T cell interactions. Following recognition of specific antigen, T cells require a second, co-stimulatory signal to induce clonal expansion and acquisition of effector function. Targeting some of these pathways is revealing more about DC biology in RA. The importance of CD40-CD40L interactions in DC and autoreactive T cell was highlighted in a study establishing the therapeutic potential of CD40 anti-sense oligonucleotides (CD40-ASO) in CIA [62]. Administered alone, CD40-ASO had no effect on progression of disease. However, encapsulation in amphoteric liposomes relieved established arthropathy, reducing CII-specific T cell responses and the production of TNFα, IL-6 and IL-17. This effect was attributed to the liposome tropism for cDCs and monocyte/macrophages. Indeed liposomal CD40-ASO efficiently inhibited CD40 expression and antigen presentation by DCs. The monoclonal antibody MR1 (specific for CD40L) efficiently abrogates histological and autoimmune aspects of CIA, highlighting the pivotal importance of this costimulatory molecule in disease pathogenesis [63].
In addition to CD40-CD40L interactions, the importance of CD80/86 interactions with CD28 and CTLA-4 in DC stimulation of T cell responses must not be ignored. The increasing mechanistic studies of abatacept function will no doubt reveal more about these costimulatory molecules.
DC THERAPEUTIC POTENTIAL
The majority of DC studies in experimental arthritis have been concerned with their use in a therapeutic context. Translation of the basic immunological principals of DC activation of T cells to the development of autoimmune arthropathy has opened avenues for manipulation of the processes involved, making reestablishment of self-tolerance in RA patients more tangible.
Repeated immunisation of mice with immature DCs [64] resulted in expansion of a regulatory T cell population capable of protecting against CIA. Adoptive transfer of these CD49b+ IL-10+ regulatory T cells was itself sufficient to prevent recipient mice developing autoimmune arthritis. CIA has also been inhibited using TNFα matured DCs [65], a surprising finding given the important proinflammatory role of this cytokine in RA, but likely exemplifying endogenous cytokine-mediated homeostasis. Indeed blockade of TNF signalling by DCs via their overexpression of soluble TNF receptor type I (sTNFRI) prevented their maturation and migration resulting in allospecific tolerance [66]. Nevertheless, vaccination with CII pulsed, TNFα matured bone marrow derived cDCs prior to induction of CIA resulted in delayed onset of disease [65]. In this instance, suppression was antigen dependent, relying on DCs having been pulsed with CII. Further study revealed that CIA inhibition by TNF treated DCs altered the effector profiles of responding T cells, switching CII induced cytokine secretion in favour of IL-5 and IL-10. Biasing of Th1/Th2 polarisation was further evidenced in the elevated ratio of anti-CII –IgG1 to –IgG2a [67].
Bone marrow derived DCs pre-treated with the anti-inflammatory cytokine IL-10 were also capable of reducing pathology in CIA, altering the Th1/Th2 bias as observed with TNF treated DCs [67]. Examination of cytokine production by both TNF- and IL-10- treated DCs revealed a reduction in IL-12p40 secretion, consistent with the lack of Th1 effector phenotypes. In contrast, dexamethasone modulated bone marrow derived DCs produced normal levels of IL-12p40, and did not skew the Th response while suppressing CIA to an equivalent degree. Examination of a variety of surface molecules related to T cell activation revealed that Dex DCs failed to up-regulate the highly conserved Delta-like 4 [68], a Notch ligand related to IFNγ production in T cells [69, 70].
While these studies have investigated the potential of DC themselves to modulate arthritis responses, exosomal products from DCs are also effective in both limiting onset and ongoing pathology in CIA. Exosomes are vesicles approximately 30-90nm in diameter produced by inner budding of the plasma membrane. These vesicles are located in the mutivesicular bodies of the late endocytic compartment of numerous cell types. Secretion of exosomes occurs when these multivesicular bodies fuse with the plasma membrane. Loading of MHC class II with exogenous peptides also occurs in these compartments with DC derived exosomes being shown to contain MHC class II costimulatory molecules CD86. Such lipid vesicles can stimulate T cells directly or through interaction with another APC and have been used to effectively stimulate anti-tumour T cell responses [71]. A series of studies have successfully ‘hijacked’ this method of intercellular communication to abrogate pathogenic responses in experimental arthritis. Bone marrow derived DCs treated with IL-10 or manipulated to overexpress IL-10, IL-4 or FasL all suppress onset of CIA and limit ongoing disease - exosomes isolated from these cells were also efficient in immunoregulation [72-76]. Peri-articular injection of FasL-overexpressing DC derived exosomes lead to localisation in CD11c+ cells at both the injection site and in the draining popliteal lymph node, providing MHC class II dependent protection from development of delayed type hypersensitivity responses [72]. Despite this effect being MHC class II dependent, exosomes from non-antigen pulsed FasL overexpressing DCs could suppress CIA. Potentially, this has both disadvantages and advantages. Lack of antigen specificity could result in unwanted immunosuppressive effects. However, if CIA suppression resulted from uptake by endogenous DCs presenting articular antigens, articular administration of such exosomes could confer protection to multiple/unknown auto-antigens, especially given that no singular inciting antigen is known to mediate RA.
The most recent innovation in this system incorporated exosomes from IDO overexpressing DCs [76]. The immuomodulatory effects of this tryptophan degrading enzyme have been well documented in experimental arthritis, with 1-MT treatment exacerbating CIA65 and DCs adenovirally transduced to express IDO preventing CIA onset and suppressing ongoing disease [76, 77]. Additionally, oral-tolerance-induced suppression of CIA generated a population of CD11c+IDO+ cells that facilitated regulatory T cell activity. Exosomes from IDO expressing DCs were found to be equally effective as the DCs themselves in suppressing established CIA [76]. Of particular note in this study was that exosomes from CTLA-4-Ig overexpressing DCs also prevented CIA by an IDO dependent mechanism. Despite elevated IDO levels in CTLA-4-Ig overexpressing DCs, the enzyme was not detected in the isolated exosomes. The authors proposed a mechanism whereby CTLA-4-Ig exosome uptake by endogenous DCs induced their expression of IDO [76], consistent with the importance of B7 ligation for IDO induction in DCs.
CONCLUSION
Models of experimental arthritis have only begun to reveal the intricacies of DC biology in RA. While manipulated DCs can be used in the suppression of experimental arthritis, the basic functions and mechanisms regarding their impact is mostly obtained indirectly by inference from other autoimmune and infectious studies. It is essential that more research into the direct contribution of DC activity in RA is forthcoming, particularly as they could hold the key to development of antigen specific therapeutics. Moreover we propose that models that minimise the effects of exogenous adjuvants will more faithfully reflect those processes that lead to the development of RA. As such they may provide fertile resource for discovery that can be translated in due course to human derived samples from early and eventually disease ‘at risk’ patient groups, increasing the likelihood of the advent of antigen specific therapeutics.