- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Cell Signaling Journal
(Discontinued)
ISSN: 1876-3901 ― Volume 4, 2012
Interplay of Bcl-2 Proteins Decides the Life or Death Fate
Grant Dewson*
Abstract
Ever since the discovery that the founding member of the Bcl-2 family of proteins contributes to tumour development by inhibiting cell survival rather than encouraging proliferation [1], interest (and controversy) in these functionally diverse homologues has raged. The Bcl-2 family of proteins participate in multiple protein-protein interactions that govern whether a cell dies in response to toxic stress. This review summarises the current knowledge of how a death stimulus culminates in the activation of Bax and Bak, the pivotal effectors of the apoptotic program, and how these critical proteins cause damage to mitochondria and the consequent demise of a cell.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 3
First Page: 3
Last Page: 8
Publisher Id: TOCELLSJ-3-3
DOI: 10.2174/1876390101103010003
Article History:
Received Date: 2/3/2010Revision Received Date: 21/8/2010
Acceptance Date: 22/10/2010
Electronic publication date: 14/4/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Walter and Eliza Hall Institute of Medical Research, Melbourne, Victoria 3052, Australia; Tel: +61 3 93452335; E-mail: dewson@wehi.edu.au
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 2-3-2010 |
Original Manuscript | Interplay of Bcl-2 Proteins Decides the Life or Death Fate | |
Characterised by conservation within Bcl-2 homology (BH) domains, the Bcl-2 proteins fall into three sub-groups (Fig. 1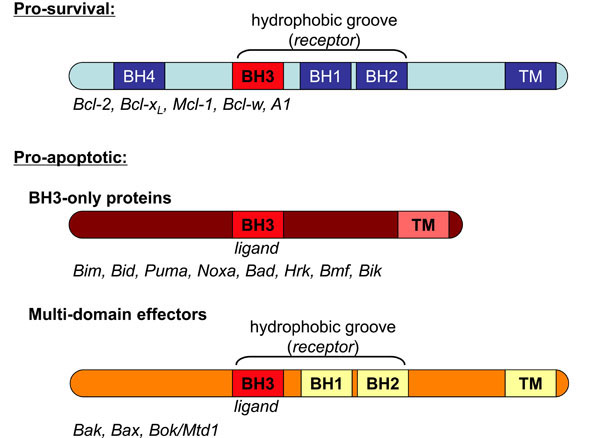 ): 1. The multi-domain pro-survival proteins, including Bcl-2, Bcl-xL, Mcl-1, A1 and Bcl-w. 2. The BH3-only proteins (e.g. Bim, Bid, Puma, Noxa, Bad), which are regulated transcriptionally or post-translationally to initiate the apoptotic response, and thereby act as the sensors of cellular damage. 3. The multi-domain pro-apoptotic effectors Bak, Bax and Bok/Mtd1.
): 1. The multi-domain pro-survival proteins, including Bcl-2, Bcl-xL, Mcl-1, A1 and Bcl-w. 2. The BH3-only proteins (e.g. Bim, Bid, Puma, Noxa, Bad), which are regulated transcriptionally or post-translationally to initiate the apoptotic response, and thereby act as the sensors of cellular damage. 3. The multi-domain pro-apoptotic effectors Bak, Bax and Bok/Mtd1.

In response to disparate death stimuli including growth factor deprivation (excellently discussed by Paul Ekert and Jabbour in this edition) and DNA damage, the interplay of the Bcl-2 proteins mediates the integrity of the mitochondrial outer membrane (MOM). Consequently an apoptotic cell is on the receiving end of a two-pronged attack. Firstly, a breached mitochondrial membrane ensures loss of oxidative phosphorylation and therefore energy production. Secondly, apoptogenic factors such as cytochrome c and Smac/DIABLO are released from the intermembrane space to activate proteolytic caspases that orchestrate destruction of the cell. Disruption of the cells power plant appears to be of paramount importance to the final outcome, because even if caspase activity is inhibited, the majority of cells with permeabilised MOM are destined to die. Bak and Bax therefore deliver the coup de grace by irreparably damaging the MOM. Despite intense investigation and numerous proposed mechanisms, how Bak and Bax deliver the death blow is unknown. Although it seems clear that the balance of pro-survival and pro-apoptotic proteins governs the life-death decision, how the complex interplay of Bcl-2 proteins allows for Bak and Bax activity to be tempered in a healthy cell and unleashed in response to cytotoxic insult is controversial. Determining how and when (or indeed if) these proteins interact is pivotal in deciphering the control of the apoptotic pathway and for potential therapeutic intervention.
BAK AND BAX- THE HARBINGERS OF DOOM
Bak and Bax functionally overlap, and can substitute to an extent in their ability to cause mitochondrial perturbation and cell death. Thus the single knock-out mice phenotypes are rather mild; bak-/- mice have no overt phenotype except modest thrombocytosis, and bax-/-mice exhibit lymphoid hyperplasia and defects in spermatogenesis [2-4] and cells from these mice are susceptible to most forms of cell death. In contrast, mice lacking both Bak and Bax die soon after birth on a mixed background [3] and cells from these mice are completely resistant to most activators of the intrinsic cell death pathway [5]. The close relative Bok/Mtd1 is less well understood. Although it cannot substitute for Bak or Bax in fibroblasts (Grant Dewson, unpublished data), Bok/Mtd1 may play a more cell type-specific Bak/Bax-like role during cell death [6, 7].
Bak and Bax adopt a similar fold as their inactive conformer (Fig. 2A ) [8,9], but despite both conserved structure and function they reside in distinct subcellular compartments in a healthy cell. Whereas Bak is constituti-vely integrated into the MOM, Bax is normally cytosolic and translocates to mitochondria only after apoptotic challenge [10]. In their native state, Bak and Bax are metastable with both undergoing structural changes to attain their activated states during cell death. Biochemical analyses have revealed activation results in significant alterations to the N-terminus, exposure of the normally buried BH3 domain and reorientation of the core a5/6 helices (Fig. 2B) [11-14]. Unfortunately, due largely to the difficulties in obtaining structures of membrane associated proteins, there is currently no high resolution structure for this activated form.
) [8,9], but despite both conserved structure and function they reside in distinct subcellular compartments in a healthy cell. Whereas Bak is constituti-vely integrated into the MOM, Bax is normally cytosolic and translocates to mitochondria only after apoptotic challenge [10]. In their native state, Bak and Bax are metastable with both undergoing structural changes to attain their activated states during cell death. Biochemical analyses have revealed activation results in significant alterations to the N-terminus, exposure of the normally buried BH3 domain and reorientation of the core a5/6 helices (Fig. 2B) [11-14]. Unfortunately, due largely to the difficulties in obtaining structures of membrane associated proteins, there is currently no high resolution structure for this activated form.
 |
Fig. (2) Bak and Bax change conformation significantly during apoptosis. A. Inactive Bak and Bax adopt a similar tertiary structure. Structures of Bak (ΔN15/ΔC25, 2IMS) and Bax (1F16) as determined by crystallography and nuclear magnetic resonance respectively [5,6]. Regions that undergo significant rearrangement during activation are indicated, α1 (blue), α2 (BH3, red), α5/6 (cyan) and α9 (TM, magenta). α helices 3-4 in combination with α5 comprise the hydrophobic groove (green), which forms the docking site for self-association and potentially for interaction with other BH3-only proteins. The Bak structure lacks both its N and C-termini. Images were generated with MacPyMOL. B. Bak/Bax conformation change during apoptosis. Bak and Bax undergo a series of conformation changes that facilitate subcellular redistribution (Bax), MOM integration and oligomerisation. Bax is normally cytosolic due to sequestration of its hydrophobic Cterminal TM domain in its hydrophobic groove (green), so that its activation involves exposure of its TM domain and translocation to and integration into the MOM. In contrast, Bak is constitutively mitochondrial via integration of its C-terminus into the MOM and therefore bypasses this redistribution phase and ensures that its hydrophobic groove is normally unoccupied. The precise order of these events is uncertain. The oligomerization of activated Bak involves insertion of its BH3 domain into the hydrophobic groove of a partner molecule. These dimers then multimerise via an interface involving the α6 helices. |
What provides the initial driving force for the transition to the activated conformation is a prevailing controversy in the field. Interaction with “activator” BH3-only proteins Bim, tBid and Puma, or with non-Bcl-2 proteins such as p53 is a potential activating mechanism [15, 16]. Alternatively, post-translational modification or changes in cellular environment such as hyperthermia or an increase in cellular pH may provide the impetus [17-19]. Once activated, Bak and Bax may interact with the inactive pool resulting in auto-activation that amplifies the apoptotic response [20, 21].
RESTRAINING BAK AND BAX PRO-APOPTOTIC FUNCTION
As Bak and Bax activity is the point of no return in an apoptotic response, its proper control is clearly crucial. Ineffectual restraint results in excessive cell death and can contribute to degenerative disorders, whilst excessive restraint inappropriately inhibits cell death and can contribute to tumourigenesis [22]. At present there are two models on curbing Bak/Bax pro-apoptotic function, termed the “direct” and the “indirect” (Fig. 3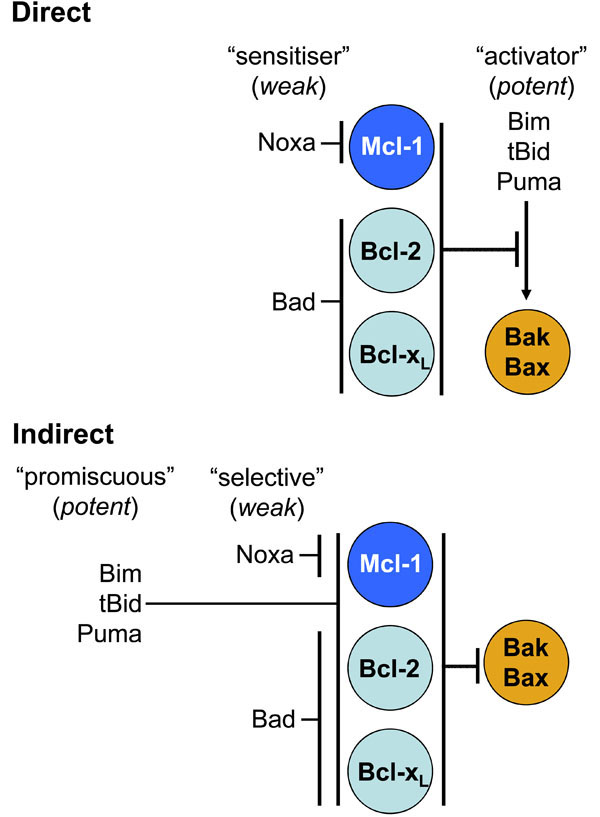 ). Neither model is entirely consistent with all available data, and aspects of both may hold [23]. The “direct” model proposes that BH3-only proteins belong to one of two distinct classes, the aforementioned “activators” (Bim, tBid and Puma) and the “sensitisers” (Bad, Noxa) [24]. The pro-survival proteins temper Bak/Bax activation by sequestering the activator BH3-only proteins. During an apoptotic response, when displaced by the sensitisers, the activators bind to Bak and Bax causing their activation [25]. However, the transient “hit and run” nature of this interaction has made it difficult to confirm in cells. This model is consistent with the greater propensity of the activator BH3-only proteins to induce cell death upon over-expression compared to the sensitiser BH3-only proteins. Inconsistent with this model is the finding that the putative activator BH3-only proteins are seemingly not required for Bax and Bak activation [26].
). Neither model is entirely consistent with all available data, and aspects of both may hold [23]. The “direct” model proposes that BH3-only proteins belong to one of two distinct classes, the aforementioned “activators” (Bim, tBid and Puma) and the “sensitisers” (Bad, Noxa) [24]. The pro-survival proteins temper Bak/Bax activation by sequestering the activator BH3-only proteins. During an apoptotic response, when displaced by the sensitisers, the activators bind to Bak and Bax causing their activation [25]. However, the transient “hit and run” nature of this interaction has made it difficult to confirm in cells. This model is consistent with the greater propensity of the activator BH3-only proteins to induce cell death upon over-expression compared to the sensitiser BH3-only proteins. Inconsistent with this model is the finding that the putative activator BH3-only proteins are seemingly not required for Bax and Bak activation [26].

Alternatively, the “indirect” model proposes that the pro-survival proteins protect cells by binding Bak/Bax. In this model the capacity of BH3-only proteins to induce apoptosis is due solely to their selective binding to pro-survival proteins [27]. Upon upregulation, the BH3-only proteins liberate Bak/Bax from pro-survival interactions allowing them to self-associate and auto-activate. The greater killing capacity of Bim, tBid and Puma may be explained by their ability to bind promiscuously with high affinity to all pro-survival proteins, rather than directly activating Bak or Bax, whilst Bad and Noxa are weaker as they selectively bind only to a subset [27]. This model is supported by the observation that Bak/Bax-dependent apoptosis can be initiated by overexpression of Noxa and Bad in combination in the absence of the activator BH3-only proteins [26]. However, in healthy cells Bax is generally regarded as monomeric, and the majority of Bak is not sequestered by pro-survival Bcl-2 proteins [28]. Furthermore, this model requires that liberated Bak or Bax already be in its active conformation or at least have undergone some conformation change. Structural analysis suggests Bak inserts its BH3 domain into the hydrophobic groove of Bcl-xL [29] (although this has not been confirmed in a complex of full-length proteins). Thus this Bak/Bax conformation at least involves exposure of the normally buried BH3 domain. So what serves as the initial activating stimulus in this case? The inherent instability of Bak and Bax may allow them to spontaneously activate, with erroneously activated Bak/Bax being restrained from self-association by binding to pro-survival proteins until inhibition is relieved upon cytotoxic stress [23]. A propensity to spontaneously activate may explain why Bak and Bax can kill a cell in the absence of the putative activator BH3-only proteins due only to sensitiser BH3-only proteins occupying the pro-survival guardians [26]. Therefore interaction with activator BH3-only proteins or post-translational modification may expedite, but may not be necessary for, Bak and Bax activation [23]. Andrews and colleagues have recently proposed a third model, the “embedded together” model, which accounts for an often neglected factor, that of the membrane localisation of the Bcl-2 proteins. This model, whilst encompassing features of both the direct and indirect models, proposes a significant role for the lipid environment of the MOM in influencing the conformation and interactions of the Bcl-2 proteins governing activation of Bak and Bax [30, 31].
Although interactions with other Bcl-2 family proteins are thought to be the dominant mode of regulation of Bak and Bax apoptotic function, interactions with non-Bcl-2 proteins have also been implicated. For example, VDAC2 (voltage-dependent anion channel 2) in the MOM binds at least a proportion of Bak in healthy cells to restrain its apoptotic function, and as a consequence, cells lacking
VDAC2 are more sensitive to death stimuli [32]. VDAC2 has also been implicated as a negative regulator of Bax under certain circumstances [33]. An emerging and contentious issue in the field is the potential role of the mitochondrial fission/fusion machinery in regulating apoptosis by modulating Bak and Bax function. Mitochondria form a dynamic network that undergoes controlled fission and fusion. These opposing processes are crucial for maintaining mitochondrial function and for the efficient segregation of mitochondria during cell division. It has been proposed that apoptosis relies on mitochondrial fission as mitochondria often fragment during apoptosis [34]. Further, the proteins that control this process have been reported to interact directly with Bax and Bak to influence their pro-apoptotic function. For example, the mitochondrial fission protein, Drp1, interacts with Bax [35]. However, a role for the fission/fusion machinery in apoptosis has been questioned as mitochondrial fragmentation is not a universal feature of apoptosis and studies investigating the role of mitochondrial fission proteins in apoptosis have provided conflicting findings (reviewed in [36]). Moreover, confirming an apoptotic role for the fission/fusion proteins (and likewise VDAC2) has been confounded by their importance for mitochondrial function and homeostasis. Nonetheless, interaction with non-Bcl-2 proteins potentially provides another layer of complexity to allow intricate control over Bak/Bax apoptotic function.
MITOCHONDRIAL PERTURBATION BY BAK/BAX OLIGOMERS
Irrespective of the activating mechanism, the resultant conformation change in Bak and Bax facilitates their self-association. Bak/Bax homo-oligomerisation is a consistent feature during apoptosis and these complexes are believed to be responsible for the damage to the MOM. Deciphering how Bak and Bax coalesce to form the killing complex is critical in determining how they permeate the MOM. We have recently shown that during apoptosis, Bak exposes its BH3 domain to insert into the hydrophobic groove of a partner molecule [12]. This symmetric BH3:groove homodimer forms the basic oligomeric subunit of the Bak and Bax pore complex [37, 38]. How this subunit nucleates to form the larger complex necessary to permeabilise the MOM is unclear but obviously must involve interfaces distinct from both the BH3 domain and groove surface [12]. Indeed, we have recently reported that during apoptosis BH3:groove homodimers of Bak subsequently multimerise via an interface involving its a6 helix thereby forming the large molecular weight complexes that may comprise the apoptotic pore [38].
Bak and Bax can potentially co-operate to kill a cell with hetero-oligomers of activated Bak and Bax detected under certain conditions [39]. Although a wild-type apoptotic response in fibroblasts lacking either Bak or Bax indicates such an association is not necessary for their pro-apoptotic function [5], it is possible that co-operativity accelerates MOM permeabilisation or sensitises certain cells (i.e. those which express limiting Bak and Bax) to apoptotic stimuli.
The exact nature of the Bak/Bax “pore” remains enigmatic with high molecular weight complexes comprising 100s of molecules detected in dying cells [40]. Whether the oligomeric complex exists as a finite proteinaceous pore or whether their aggregation non-specifically destabilises the lipid bilayer is disputed, although the concomitant release of a plethora of intermembrane space proteins of varying sizes during cell death may be more consistent with non-specific membrane disruption. At least in artificial membranes, activated Bak or Bax can permeabilise membranes without assistance from other proteins. In cells however a variety of mitochondrial proteins have been proposed to assist Bak/Bax in MOM permeabilisation. For example, hi-jacking the aforementioned mitochondrial fission/ fusion machinery via association of Bak/Bax with Drp1 and mitofusins [35].
Structural homology of Bcl-2 proteins to the pore-forming domains of bacterial colicins and diphtheria toxin hints at a potential mechanism for membrane perturbation [41]. The pore-forming domain theory posits that mitochondrial perturbation is due to the hydrophobic a5/6 helices of Bax/Bak inserting into the MOM. Indeed Andrews and colleagues have elegantly shown membrane insertion of a5/6 of Bax, and intriguingly also Bcl-2, in response to an apoptotic stimulus [11, 42]. The pore-forming toxins are thought to form lipidic “toroidal” pores, whereby lipid bilayer curvature results in a solvent exposed pore surface comprised of both a-helical peptides and phospholipid headgroups. Bcl-2 homologues are reported to induce similar membrane curvature [43, 44], with activated Bax (or peptides derived from Bax) shown to induce a lipid pore structure [45, 46]. As both pro-apoptotic Bax and pro-survival Bcl-2 similarly insert their a5/6 into MOM, what distinguishes a pro-apoptotic protein from a pro-survival protein is unclear, but presumably relates to their ability to oligomerise and nucleate a “pore”. As a5 forms the backbone of the hydrophobic groove of Bak and Bax, its insertion into the MOM may disrupt this critical binding surface. Therefore what role BH3:groove-mediated oligomerisation plays in a5/6 insertion is currently unclear.
ITS A GROOVE THING
While it is clear that the intricate interplay of the Bcl-2 family of proteins exerts critical control over whether a cell lives or dies, controversy still reigns regarding what the crucial interactions are and when they occur. There is substantial evidence that the interplay of Bcl-2 proteins occurs via BH3:groove associations, including structural and binding studies that show pro-survival proteins are able to bind BH3-only proteins, as well as Bak and Bax in this way. We have also shown that Bak self-association also involves a BH3:groove interaction [12]. As the hydrophobic groove of Bak/Bax can act as a receptor surface for BH3 domains, insertion of the BH3 domain from activator BH3-only proteins into the groove of Bak/Bax is therefore a plausible mechanism for direct activation. As Bak/Bax activation involves exposure of their BH3 domain this is likely to disrupt the binding groove and thereby provide a molecular mechanism for displacing the BH3-only protein as proposed by the “hit and run” model. Somewhat surprisingly, a recent study showed that a modified BH3 peptide from Bim initially binds to Bax a-helices 1 and 6, rather than its groove [47]. Whether such an interface is retained in full-length proteins, is conserved for direct activation of Bak, and how this binding signal is subsequently translated into the Bak/Bax structural alterations outlined in Fig. (1) is of intense interest.
Insight into the molecular interactions of the Bcl-2 homologues is critical in understanding how the apoptotic machinery is regulated and therefore how it can be manipulated for therapeutic benefit. The recent development of BH3 mimetic compounds that interfere with the interactions between the Bcl-2 proteins heralds a new era for targeted cancer therapy. Conservation of the BH3 domain:groove interaction highlights the need for a clear understanding of the molecular interplay of these critical proteins. The myriad of associations ultimately regulates Bak/Bax activity and thus commitment to death. Elucidating the structure of Bak/Bax as their activated, oligomerised conformer, importantly in the context of a membrane, will finally reveal how Bak and Bax permeabilise the MOM and assert their deadly influence.
ACKNOWLEDGEMENTS
GD is supported by grants from the National Health and Medical Research Council Australia and Association for International Cancer Research.