- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Condensed Matter Physics Journal
(Discontinued)
ISSN: 1874-1924 ― Volume 4, 2013
Theoretical Study of the Effect of Pressure on the Electronic Structure of Grey Tin
Mohammad G. Merdan1, Mudar A. Abdulsattar 2, Ahmed M. Abdul-Lettif1, *
Abstract
A large unit cell (LUC) within intermediate neglect of differential overlap (INDO) formalism has been used to estimate the electronic properties of α - Sn and their pressure dependence. The calculated properties are, in general, in acceptable agreement with the available experimental values except the direct band gap. The calculated properties follow the usual qualitative trends that can be obtained from Hartree-Fock analysis. The increase of pressure is predicted to cause: a linear increase of the direct band gap with a pressure coefficient of 0.06 eV/GPa, a linear increase of the valence band width, a decrease of the conduction band width, a slight decrease of the electronic occupation probability for the s orbital with a slight increase of this probability for the p orbital, and a decrease of the X-ray scattering factors.
Article Information
Identifiers and Pagination:
Year: 2009Volume: 2
First Page: 30
Last Page: 36
Publisher Id: TOCMPJ-2-30
DOI: 10.2174/1874186X00902010030
Article History:
Received Date: 12/2/2009Revision Received Date: 2/6/2009
Acceptance Date: 10/6/2009
Electronic publication date: 16/07/2009
Collection year: 2009
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/ which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the College of Science, Babylon University, Hilla, Iraq; E-mail: abdullettif@yahoo.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 12-2-2009 |
Original Manuscript | Theoretical Study of the Effect of Pressure on the Electronic Structure of Grey Tin | |
1. INTRODUCTION
The electronic and structural properties of tin are very sensitive to pressure conditions [1]. The ground state, grey tin (α - Sn), has the diamond structure and is known to be a semiconductor with zero band gap (semi-metal). Tin also exists in the β - Sn structure (white tin) at atmospheric pressure above 13oC. The metallic β - Sn phase is a tetragonal distortion of diamond with two atoms per unit cell. This phase is stable up to 9.5 GPa at room temperature, where it transforms to a bct form, followed by a transformation to the cubic bcc structure [2]. These temperature and pressure-driven phase transformations have caused tin to be of considerable experimental and theoretical interest [2-1].
The electronic and structural properties of complex systems like tin require a fully quantum-mechanical description. Accurate full first-principles calculations such as Hartree-Fock with correlation correction [12] and the local density approximation [13, 14] are extremely demanding from the computational point of view. The development of simpler yet reliable approximate methods of calculations is therefore crucial to progress in this field.
An alternative to first principle methods, which has already provided numerous significant results, is the so called the large unit cell-intermediate neglect of differential overlap (LUC-INDO) [15-18]. The semi-empirical LUC-INDO method is capable of simulating real crystals because it takes into account the property of periodicity. Due to its semi-empirical character and a specific parameterization scheme, the computer program is not cumbersome and time consuming (about 1 hour for each process cycle) in the treatment of the electronic and spatial structure of simple systems as in our case. Also, it is suitable for analyzing complex systems [19-22].
A lot of studies have been carried out on the properties of tin and its phase transitions [1-1]. However, there are comparatively fewer studies on the pressure and temperature dependence of these properties. The aim of the present work is to investigate the effect of pressure on the electronic and structural properties of α - Sn using the LUC-INDO formalism which will be outlined in the next section.
2. CALCULATION METHOD
A quantum-chemical semi-empirical INDO method developed especially for crystals [23] is used in the present work. This quantum computational formalism has been used with great success especially exploiting the so-called LUC (Large Unit Cell) model [18]. Within the method each molecular orbital is constructed as a linear combination of atomic orbitals [23] in order to express the wave function of the system. Each energy value is calculated by the HF self-consistent field method and the total energy of the system is obtained. The basic idea of LUC is in computing the electronic structure of the unit cell extended in a special manner at k=0 in the reduced Brillouin zone. This equivalent to a band structure calculation at those k points; which transform to the Brillouin zone center on extending the unit cell [18].
The crystal wave function in the LUC-INDO formalism is written in the following form:
where NL is the number of LUCs in the crystal, Cp(k) denotes the combination coefficients of the hybridized orbitals, φ is the atomic wave function, k is the wave vector, and Tv denotes the lattice translation vector. The Hartree-Fock equation in this case ( Roothaan-Hall equation ) can be written as [24]:
where the summation index p goes over all the atomic states of the LUC, Fpq(k) represents the Fock Hamiltonian which is given by
and Spq(k) is the overlap integral defined by
where HT in Eq.(3) represents the Hamiltonian operator of the total energy ET which is defined as
where the first term of the right side of Eq.(5) represents the inter-nuclear potential energy, and Pλσis the density matrix which has the following expression:
The summation is over the occupied (occ) orbitals only.
where ZAis the core charge, and the summation is over all nuclei. It should be pointed out that our calculations are carried out at k=0, so k value in eqs.(1,3,4,6) is set to zero. For more details of LUC-INDO formalism and the final form of the Fock matrix elements at k=0, see Refs [15-18, 23-26]. The Roothaan-Hall equation are solved by first assuming an initial set of the linear expansion coefficient (Cp(k)), generating the density matrix (Pλσ), and computing the overlap integral and the first guess of the Fock matrix elements (Fpq(k)). Then one can calculate the electronic energy (Ee), and a new matrix of Cp(k) coefficients can be obtained. This procedure is continued until there is no significant variation between the calculated value of Cp(k) and E of the successive iterations.
An initial guess of the wave function is predicted using the basis set and adopting Slater-type orbitals [23]. The initial guess of the wave function is important since an optimum guess reduces the number of iterations performed to obtain the converged electronic energy. A large number of iterations will result in an accumulation of the computational errors. The sp3 initial guess of the wave function is given as an expected linear combination of the atomic states of one cell. The tolerance of convergence of the total electronic energy adopted in our calculations is 10-8 eV. A large unit cell of 8 atoms, which is the conventional Bravais lattice of the diamond structure, has been used. Interactions of the atoms in the central Bravais lattice with the surrounding atoms up to the fourth neighbors are included. It should be pointed out that the increase of the LUC size will result in an increase of the results accuracy [27], but this complicates the calculations and needs very long time in comparison with the time needed for 8-atom LUC calculations.
3. RESULTS AND DISCUSSION
3.1. Choice of Empirical Parameters
The empirical parameters included in the LUC-INDO method are the orbital exponent ζ, the bonding parameter
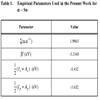
3.2. Electronic and Structural Properties
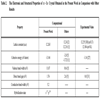
Using the computational procedure described in section 2, the electronic and structural properties of α - Sn crystal at 0 K and zero pressure are calculated as listed in Table 2 in comparison with other computational and experimental results.
The equilibrium lattice constant (a0) is determined by plotting the total energy as a function of the lattice parameter, as depicted in Fig. (1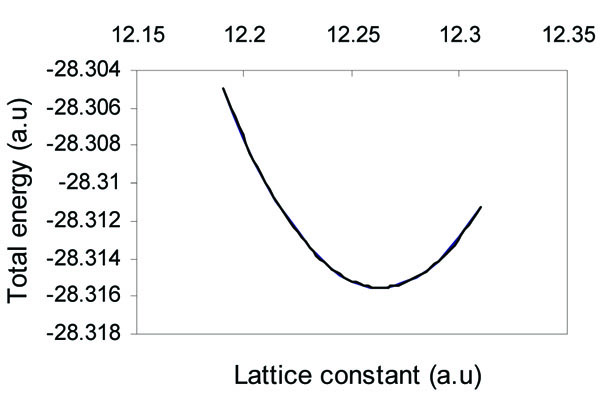 ). The calculated value of the equilibrium lattice constant (12.263 a.u) is in good agreement with the estimated value of 12.184 a.u from the experimental data [30].
). The calculated value of the equilibrium lattice constant (12.263 a.u) is in good agreement with the estimated value of 12.184 a.u from the experimental data [30].
 |
Fig. (1) Total energy of (α - Sn) as a function of lattice constant. |
The cohesive energy is calculated from the total energy of the large unit cell. Since the large unit cell in the present work is composed of 8 atoms, the cohesive energy can be determined from the following expression:
where Efree denotes the free atom sp shell energy, E0 is the vibrational energy state, which is referred to as zero-point energy, and its value is 0.03 eV [32]. The cohesive energy value of present work (-3.144 eV) is also in good agreement with the experimental value of -3.14 eV [31]. This is due to the including of the exchange integrals, correlation correction, and zero-point energy in the present analysis. The correlation correction is the correction included to take into account the fact that the motions of electrons are correlated pairwise to keep electrons apart. Correlation energies may be included by considering Slater determinants composed of orbitals that represent excited state contribution, and this method of including unoccupied orbitals in the many body wavefunction is referred to as configuration interaction (CI) [24]. Another method to estimate the correlation correction, which is used in the present work, is the Moller-Plesset perturbation theory [24]. This correction is calculated to be 0.27 eV. A detailed description of the several kinds of correlation corrections is present in reference [24].
The calculated direct band gap is considerably larger than the experimental value. A similar anomaly was also obtained by Svane [5] using the Hartree-Fock approximation. The large computed value of the direct band gap can be attributed to the approximations involved in the LUC-INDO formalism and Hartree-Fock method, and to the perturbative treatment of the correlation correction [33]. However, the available non-perturbative correlation correction method takes nearly ten times the computational time needed in Moller-Plesset method. The most significant approximations that affect the band gap are:
- Using equal values of ζ and β° for s and p wavefunctions. The difference between bonding and anti-bonding states is directly proportional to β°S, where the overlap integral S is a function of ζ. The percentage different of ζ between s and p orbitals was reported to be 14.4% for tin [28].
- Neglecting the core states will also result in a neglect of its effects on the distribution of the outer valence electrons.
- Using the 5s 5p orbitals only without the inclusion of the 4d orbital.
It should be pointed out that the present value for the band gap is a non-relativistic value. The computed band gap value of tin is expected to be in good agreement with the corresponding experimental value if the relativistic splitting of the levels [5] is considered, and if greater size of LUC is used [34], in addition to the points mentioned above.
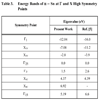
The eigenvalues of the high symmetry points used to determine the band structure are listed in Table 3, in comparison with the corresponding values calculated by Svane [5].
Some physical properties of solids can be determined using the analysis described in the preceding section, such as the electronic charge density and the X-ray scattering factor, the valence electrons charge density (ρe(r)) can be expressed as:
The electronic charge density for some planes of α - Sn crystal is displayed in Fig. (2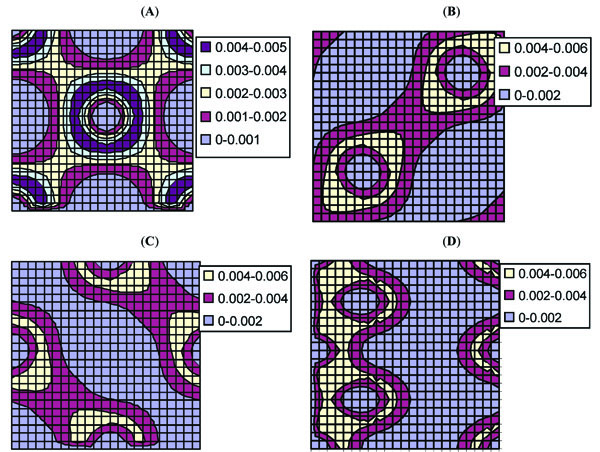 ).
).
 |
Fig. (2) Valence charge density (in atomic unit) of α - Sn in the: (A) (001) plane, (B) (400) plane, (C) (200) plane, and (D) (110) plane. |
The X-ray scattering factor (fj) is defined by [31].
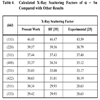
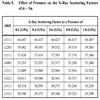
where G is the reciprocal lattice vector. In Table 4, the calculated X-ray scattering factors for α - Sn are listed in comparison with other computational [35] and experimental [25] results. The calculated X-ray scattering factors are in good agreement with the experimental values.
3.3. Effect of pressure on the properties of α - Sn
The pressure dependence of the electronic structure can be predicted from the present computational analysis. The pressure dependence of the lattice parameter is determined using the Murnaghan [36] equation of state:
where a is the lattice parameter at pressure P, ao is the lattice constant at zero pressure, Bo is the bulk modulus at zero pressure, and 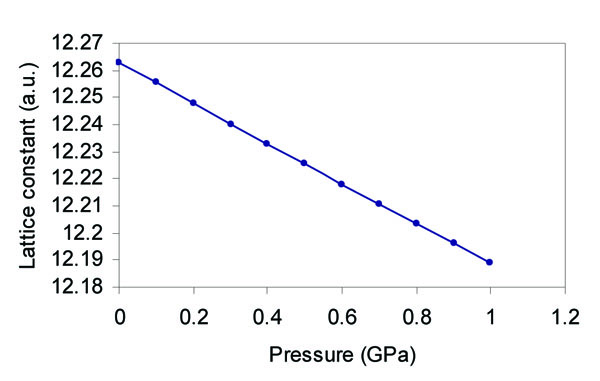 ). This figure was plotted using the experimental value of B0 of 53 GPa [38] and the calculated value of
). This figure was plotted using the experimental value of B0 of 53 GPa [38] and the calculated value of
 |
Fig. (3) Lattice constant of (α - Sn) as a function of pressure. |
The pressure dependence of the cohesive energy, direct band gap, valence band width, and conduction band width as predicted from our analysis is shown in Figs. (4 -7
-7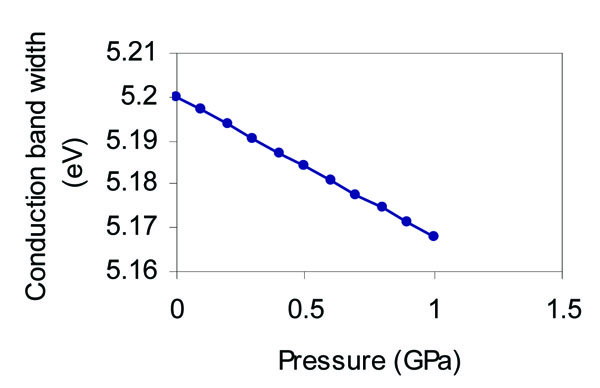 ) respectively.
) respectively.
 |
Fig. (4) Effect of pressure on the cohesive energy of grey tin. |
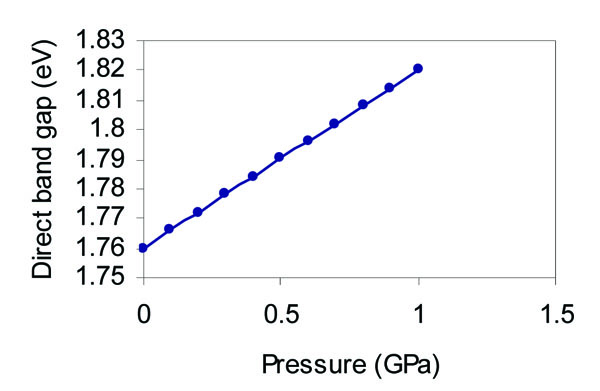 |
Fig. (5) Effect of pressure on the direct band gap width of grey tin. |
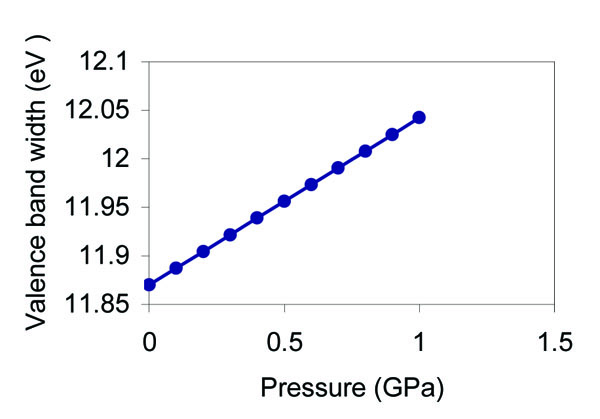 |
Fig. (6) Effect of pressure on the valence band width of grey tin. |
 |
Fig. (7) Effect of pressure on the conduction band width of grey tin. |
As it is obvious from Fig. (5 ), the direct band gap increases nearly linearly with the increase of the pressure. We suppose that this is mainly due to the nearly linear dependence of the lattice constant with the pressure as shown in Fig. (3), and since the translation vector (Tv) scales linearly with the lattice constant, one can expect a nearly linear relation between the band gap and the pressure according to the present model. The pressure derivative of the direct band gap equals 0.06 eV/GPa. Wei and Zunger [37] have determined the pressure derivative of the band gap of the three main transitions; Г- X, Г- L, and Г- Г for α - Sn to be -0.01, 0.059, and 0.166 eV/GPa respectively. The direct band gap is determine, d from the energy difference (Г2-Г25). The observed linear increase of the direct band gap with pressure is due to the linear increase of the Г2 energy with respect to the Г25 energy as found from the present analysis, and also by Bassani and Liu [3]. The valence band width is calculated from the energy difference (Г25-Г1). The Г1 state is shown to decrease linearly with the increase of pressure, and this causes a linear increase of the valence band width as depicted in Fig. (6
), the direct band gap increases nearly linearly with the increase of the pressure. We suppose that this is mainly due to the nearly linear dependence of the lattice constant with the pressure as shown in Fig. (3), and since the translation vector (Tv) scales linearly with the lattice constant, one can expect a nearly linear relation between the band gap and the pressure according to the present model. The pressure derivative of the direct band gap equals 0.06 eV/GPa. Wei and Zunger [37] have determined the pressure derivative of the band gap of the three main transitions; Г- X, Г- L, and Г- Г for α - Sn to be -0.01, 0.059, and 0.166 eV/GPa respectively. The direct band gap is determine, d from the energy difference (Г2-Г25). The observed linear increase of the direct band gap with pressure is due to the linear increase of the Г2 energy with respect to the Г25 energy as found from the present analysis, and also by Bassani and Liu [3]. The valence band width is calculated from the energy difference (Г25-Г1). The Г1 state is shown to decrease linearly with the increase of pressure, and this causes a linear increase of the valence band width as depicted in Fig. (6 ). The conduction band width can be determined from the energy difference (X4c-Г2). Although the X4C energy is shown to increase with pressure, the conduction band width decreases with pressure as shown in Fig. (7) because the increase of Г2 energy is more than the increase of X4C energy.
). The conduction band width can be determined from the energy difference (X4c-Г2). Although the X4C energy is shown to increase with pressure, the conduction band width decreases with pressure as shown in Fig. (7) because the increase of Г2 energy is more than the increase of X4C energy.
It is found that the s state occupation decreases with the increase of pressure as in Fig. (8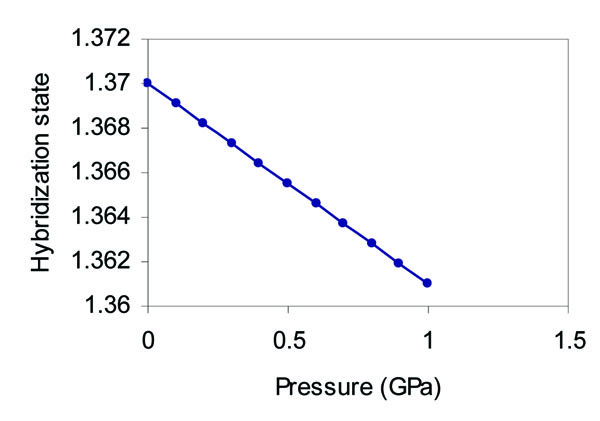 ), whereas the p state occupation increases with the increase of pressure as in Fig. (9
), whereas the p state occupation increases with the increase of pressure as in Fig. (9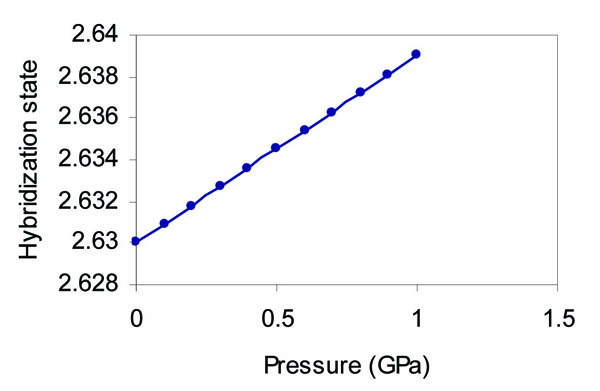 ).
).
 |
Fig. (8) Effect of pressure on the hybridization state of the s orbital of grey tin. |
 |
Fig. (9) Effect of pressure on the hybridization state of the p orbital of grey tin. |
The present works shows that the X-ray scattering factors decrease as pressure increases. This behavior is obvious in Table 5. This can be interpreted as follows: increasing pressure decreases the inter-planer distance (dhkl), this increases the Bragg scattering angle (Bragg's law), and this in turn causes a decrease of the scattering wave intensity.
4. CONCLUSIONS
The present work shows that the semi-empirical LUC-INDO model can be used to simulate solids in a practical manner, because it is reasonably economical to use. The success of the approach depends on the optimum choice of the empirical parameter set and on the size of the LUC. Increasing the LUC size is expected to improve the result accuracy and reliability as it was confirmed by Harker and Larkins [27], but the increase of the LUC size results in a significant increase of the processing time. The calculated properties of α - Sn are, in general, in acceptable agreement with the available experimental values, except the direct band gap. Increasing pressure is predicted to cause the following effects; a decrease of the absolute value of the cohesive energy, a linear increase of the direct band gap with a pressure coefficient of 0.06 eV/GPa, a linear increase of the valence band width, a decrease of the conduction band width, a slight decrease of the electronic occupation probability for the s orbital with a slight increase of this probability for the p orbital, and a decrease of the X-ray scattering factors. The above results show the usual trends that can be obtained from Hartree-Fock analysis which are a high band gap and acceptable qualitative results.
ACKNOWLEDGEMENTS
The present work was supported in part by the College of Science in Babylon University. The authors would like to thank Miss Suha Taha for here technical efforts.