- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Pediatric Medicine Journal
(Discontinued)
ISSN: 1874-3099 ― Volume 10, 2020
Infantile GM1-Gangliosidosis Revealed by Slate-Grey Mongolian Spots
Ghizlane Draïss1, Adil Fouad1, *, Nourddine Rada1, Ouafa Hocar2, Naima Fdil3, Mohamed Bouskraoui1
Abstract
Introduction:
GM1-gangliosidosis is an inherited metabolic disease caused by mutations in the GLB1 gene resulting in deficiency of β-galactosidase. Three forms have been identified: Infantile, juvenile, and adult. The infantile type progresses rapidly and aggressively and a delayed diagnosis hampers the prevention of many neurological deficits. This delay in diagnosis may be due to the variability of clinical expression of the disorder.
Hypothesis:
Extensive Mongolian or slate-grey spots deserve special attention as possible indications of associated inborn errors of metabolism, especially GM1-gangliosidosis and mucopolysaccharidosis. Only symptomatic treatments are available for GM1-gangliosidosis; research is underway.
Observation:
In this article, we report a case of infantile GM1-gangliosidosis revealed by slate-grey Mongolian spots, a rare condition in Morocco, and a review of the literature.
Conclusion:
The finding of persistent and extensive slate-grey mongolian spots in infant could lead to early detection of GM1-gangliosidosis before irreversible organ damage occurs.
Article Information

Identifiers and Pagination:
Year: 2019Volume: 9
First Page: 1
Last Page: 4
Publisher Id: TOPEDJ-9-1
DOI: 10.2174/1874309901909010001
Article History:
Received Date: 2/8/2018Revision Received Date: 19/11/2018
Acceptance Date: 20/12/2018
Electronic publication date: 31/1/2019
Collection year: 2019
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: (https://creativecommons.org/licenses/by/4.0/legalcode). This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
* Address correspondence to this author at the Department of Pediatrics, Mohamed VI University Hospital, PB 2360, Ibn Sina Avenue, Marrakesh, Morocco; Tel: +212524300700; E-mail: adilfouad@hotmail.fr
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 2-8-2018 |
Original Manuscript | Infantile GM1-Gangliosidosis Revealed by Slate-Grey Mongolian Spots | |
1. INTRODUCTION
Gangliosidosis are a group of autosomal recessive inherited metabolic disorders, in which accumulation of gangliosides in the CNS leads to severe and progressive neurological impairment. GM1-gangliosidosis is caused by mutations in the GLB1 gene, resulting in the deficiency of β-galactosidase and subsequent accumulation of GM1-ganglioside that accumulates in the brain as well as in the liver, spleen and kidney. Clinically, GM1-gangliosidosis can be classified into three types, type I (infantile), type II (juvenile) and type III (adult). Type I shows a rapid clinical course at the onset of the infantile period and is characterized clinically by neurological symptoms, bone abnormalities and hepatosplenomegaly [1Ida H, Eto Y, Maekawa K. Fetal GM1-gangliosidosis: Morphological and biochemical studies. Brain Dev 1989; 11(): 394-8.
[http://dx.doi.org/10.1016/S0387-7604(89)80023-3] , 2Lee JS, Choi JM, Lee M, et al. Diagnostic challenge for the rare lysosomal storage disease: Late infantile GM1-gangliosidosis. Brain Dev 2018; 40(5): 383-90.
[http://dx.doi.org/10.1016/j.braindev.2018.01.009] [PMID: 294 39846] ]. Mongolian or slategrey spots are grey-blue, congenital pigment spots that suggest an accumulation of dopapositive melanocytes. They are usually round or ovoid, single or multiple and occur in the sacrococcygeal region. Classically, benign and limited, they appear at birth or in the first months of life and disappear spontaneously before puberty. However, when extensive, they deserve special attention. An association between Mongolian spots and inborn errors of metabolism, especially GM1-gangliosidosis and mucopolysaccharidoses, has been described [3Abilkassem R, Agadr A. Tache mongoloïde extensive: Un signe clinique qui mérite une attention particulière. Pan Afr Med J 2013; 16: 41.
[http://dx.doi.org/10.11604/pamj.2013.16.41.3062] [PMID: 246 48854] ]. We present a case of infantile GM1-gangliosidosis revealed by slate-grey spots, a rarely described disease in Morocco, and a review of the literature.
2. CASE REPORT
The patient, a 7-month-old male infant, the only living son of parents of a second degree consanguineous marriage, was born at full-term and adapted well to extrauterine life. His psychomotor development was normal until the age of 4 months (gesticulated normally, smiled, followed light). An older brother had died at the age of 6 months due to respiratory distress with a clinical picture of psychomotor retardation, macrocephaly, abdominal distension, and blue-grey pigmented cutaneous spots. The present patient was hospitalized (1st hospitalization) for respiratory distress with the appearance of cutaneous spots and psychomotor regression since the age of 4 months. He had coarse facial features, macrocephaly (cranial circumference + 3DS), abolished tendon reflexes, significant psychomotor retardation (axial hypotonia, no voluntary prehension, no smile, sitting position even with unearned support), slate-grey spots scattered on the trunk and limbs, a negative Darier sign, and some atrophic lesions on the trunk (Fig. 1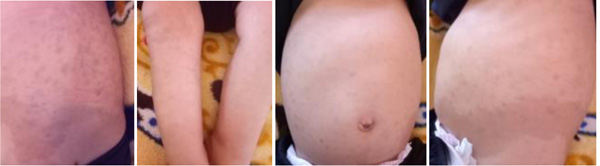 ). The ophthalmological examination suggested bilateral blindness (did not follow light), a pale papilla, but no cherry-red macular spot. He had no skeletal deformities. Brain imaging showed matter myelination abnormalities, enlargement of frontotemporal subarachnoid space and hypoplasia of the corpus callosum (Fig. 2
). The ophthalmological examination suggested bilateral blindness (did not follow light), a pale papilla, but no cherry-red macular spot. He had no skeletal deformities. Brain imaging showed matter myelination abnormalities, enlargement of frontotemporal subarachnoid space and hypoplasia of the corpus callosum (Fig. 2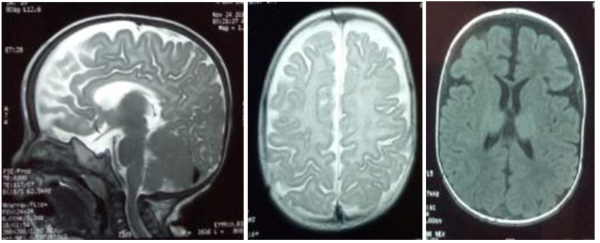 ). Abdominal ultrasonography revealed homogeneous hepatosplenomegaly. He had no atypical or vacuolated lymphocytes on the blood smear, liver and thyroid tests were normal, the amino acid chromatography was normal and the urinary excretion of glycosaminoglycans was negative. The presence of oligosaccharides in the urine, and serum beta-galactosidase deficiency confirmed the diagnosis of GM1-gangliosidosis. The progression was marked by death at the age of 9 months after severe neurological deterioration. The infant died before genetic testing could be performed and genetic counselling was planned for the parents.
). Abdominal ultrasonography revealed homogeneous hepatosplenomegaly. He had no atypical or vacuolated lymphocytes on the blood smear, liver and thyroid tests were normal, the amino acid chromatography was normal and the urinary excretion of glycosaminoglycans was negative. The presence of oligosaccharides in the urine, and serum beta-galactosidase deficiency confirmed the diagnosis of GM1-gangliosidosis. The progression was marked by death at the age of 9 months after severe neurological deterioration. The infant died before genetic testing could be performed and genetic counselling was planned for the parents.
 |
Fig. (1) Pictures of slate-grey spots on the trunk and limbs. |
 |
Fig. (2) MRI scans showing matter myelination abnormalities, enlargement of the fronto-temporal subarachnoid space, and hypoplasia of the corpus callosum. |
3. DISCUSSION
GM1-gangliosidosis is a neurodegenerative condition for which three main clinical forms have been identified: Type I (severe), type II, and type III. The incidence of GM1-gangliosidosis is considered to be between 1/100. 000 and 1/200.000 live births. The infantile type is characterized by psychomotor regression by the age of 6 months, visceromegaly, cherry-red macular spots with facial and skeletal abnormalities. In some patients with the infantile form, cardiomyopathy may sometimes be present. It is thought that impaired elastogenesis and cardiomyopathy in GM1-gangliosidosis are caused by defects in the elastinbinding domain of elastinbinding protein [4Sperb F, Vairo F, Burin M, Mayer FQ, Matte U, Giugliani R. Genotypic and phenotypic characterization of Brazilian patients with GM1-gangliosidosis. Gene 2013; 512(1): 113-6.
[http://dx.doi.org/10.1016/j.gene.2012.09.106] [PMID: 23046 582] , 5Yang CF, Wu JY, Tsai FJ. Three novel beta-galactosidase gene mutations in Han Chinese patients with GM1-gangliosidosis are correlated with disease severity. J Biomed Sci 2010; 17: 79.
[http://dx.doi.org/10.1186/1423-0127-17-79] [PMID: 209202 81] ]. Juvenile GM1-gangliosidosis usually starts between 7 months and 3 years of age with slowly progressing neurological signs including early motor problems, strabismus, muscle weakness, seizures, lethargy, and susceptibility to infections. The adult form, the attenuated phenotype, with the onset between 3 and 30 years, is characterized by cerebellar dysfunction, dystonia, slurred speech, short stature and mild vertebral deformities [6Armstrong-Javors A, Chu CJ. Child neurology: Exaggerated dermal melanocytosis in a hypotonic infant: A harbinger of GM1-gangliosidosis. Neurology 2014; 83(17): e166-8.
[http://dx.doi.org/10.1212/WNL.0000000000000912] [PMID: 253324 52] , 7Caciotti A, Garman SC, Rivera-Colón Y, et al. GM1-gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim Biophys Acta 2011; 1812(7): 782-90.
[http://dx.doi.org/10.1016/j.bbadis.2011.03.018] [PMID: 21497194] ].
Among different ethnic groups, over 90% of Native Americans and people of African descent, approximately 80% of Asians, about 70% of Hispanics, and fewer than 10% of Caucasians have Mongolian spots [8Ashrafi MR, Shabanian R, Mohammadi M, Kavusi S. Extensive Mongolian spots: A clinical sign merits special attention. Pediatr Neurol 2006; 34(2): 143-5.
[http://dx.doi.org/10.1016/j.pediatrneurol.2005.07.010] [PMID: 1645882 9] ]. Typical Mongolian spots are limited benign blue or slate grey macular lesions of varying size, most commonly located in the lumbosacral region. Mongolian spots usually disappear gradually with age, but may occasionally persist. Those associated with inborn errors of metabolism are extensive and show no signs of resolution. Extensive Mongolian spots are most frequently associated with GM1-gangliosidosis, Hurler-Scheie syndrome, Niemann-Pick disease, Hunter syndrome and α-mannosidosis. The findings of generalized Mongolian spots in an infant may represent underlying storage disorders thereby allowing identification of families at risk. They may also take on a much darker nuance in colour [9Kumar Bhardwaj N, Khera D. Mongolian spots in GM1-Gangliosidosis. Indian Pediatr 2016; 53(12): 1133.
[PMID: 28064276] , 10Mimouni-Bloch A, Finezilber Y, Rothschild M, Raas-Rothschild A. Extensive Mongolian Spots and Lysosomal Storage Diseases. J Pediatr 2016; 170: 333-e1.
[http://dx.doi.org/10.1016/j.jpeds.2015.11.009] [PMID: 267038 76] ]. Mongolian spots result from entrapment of melanocytes in the dermis because of arrested transdermal migration from the neural crest into the epidermis. The accumulated metabolites in GM1-gangliosidosis are thought to bind tightly to tyrosine kinase protein, resulting in an increased nerve growth factor. This factor then binds to chemotactic melanocyte receptors, leading to abnormalities in melanocyte migration and producing the large dermal melanocytosis seen [11Hackbart BA, Arita JH, Pinho RS, Masruha MR, Vilanova LCP. Mongolian spots are not always a benign sign. J Pediatr 2013; 162(5): 1070.
[http://dx.doi.org/10.1016/j.jpeds.2012.12.044] [PMID: 23374 678] ]. Demonstration of decreased betagalactosidase activity in cultured skin fibroblasts, urine, white blood cells or amniotic fluid cells establishes the diagnosis [12Weissbluth M, Esterly NB, Caro WA. Report of an infant with GM1-gangliosidosis type I and extensive and unusual mongolian spots. Br J Dermatol 1981; 104(2): 195-200.
[http://dx.doi.org/10.1111/j.1365-2133.1981.tb00045.x] [PMID: 678306 1] ]. Our patient had facial dysmorphism, hepatosplenomegaly, and slategrey dermal spots suggesting a storage disease. Results of a skin biopsy were inconclusive but a urinary metabolite investigation revealed oligosaccharides suggestive of GM1-gangliosidosis, subsequently confirmed by enzymatic assay.
There are as yet no approved treatments for GM1-gangliosidosis. Research is underway in animal models evaluating gene therapy technologies and intravenous enzyme replacement therapies [13Jarnes Utz JR, Kim S, King K, et al. Infantile gangliosidoses: Mapping a timeline of clinical changes. Mol Genet Metab 2017; 121(2): 170-9.
[http://dx.doi.org/10.1016/j.ymgme.2017.04.011] [PMID: 2847 6546] ]. At present, only symptomatic treatments are available. Substrate reduction therapy mediated by N-butyldeoxynojirimycin (NB-DNJ, Miglustat) has been used with encouraging results for the treatment of patients with mild to moderate type 1 Gaucher disease, Niemann-Pick disease type C and Sandhoff type chronic GM2 gangliosidosis [7Caciotti A, Garman SC, Rivera-Colón Y, et al. GM1-gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim Biophys Acta 2011; 1812(7): 782-90.
[http://dx.doi.org/10.1016/j.bbadis.2011.03.018] [PMID: 21497194] ].
Substrate reduction therapy using miglustat has been tried in the infantile gangliosidosis patient population (both GM1 and GM2-gangliosidoses). It has been generally well tolerated and has been shown to be safe in this population. It has not been observed to result in marked improvement in symptom management or disease progression. Safety of low-carbohydrate diets in an infantile patient group, however, may be of concern in terms of maintaining adequate nutrition [13Jarnes Utz JR, Kim S, King K, et al. Infantile gangliosidoses: Mapping a timeline of clinical changes. Mol Genet Metab 2017; 121(2): 170-9.
[http://dx.doi.org/10.1016/j.ymgme.2017.04.011] [PMID: 2847 6546] ]. Intracranial delivery of recombinant Adeno-Associated Virus (AAV) vectors has been shown to be highly effective in animal models. The choice of injection sites and promoters driving transgene expression are important parameters to translate into clinical trials a safe and effective AAV-based therapy for GM1-gangliosidosis [14Stoica L, Todeasa S, Lata J, Martin D, Seana-Esteves M. Optimization of AAV-gene therapy for GM1-gangliosidosis. Mol Ther 2016; 24: 149.
[http://dx.doi.org/10.1016/S1525-0016(16)33182-3] ]. However, some researchers find that cyclodextrins are useful for the treatment of GM1-gangliosidosis [15Yuki M, Keiichi M, Taishi H, et al. Feasibility study of cyclodextrins as active pharmaceutical ingredients for the treatment of GM1-gangliosidosis. Asian journal of pharmaceutical sciences 2016; 11: 183-4.]. On the other hand, other researchers find that N-Functionalized amino (hydroxymethyl) cyclopentanetriols are potent inhibitors of β-D-galactosidases and, for the first time, could be shown to act as pharmacological chaperones for GM1-gangliosidosis-associated lysosomal acid β-galactosidase thus representing a new structural type of pharmacological chaperones for this lysosomal storage disease [16Schalli M, Weber P, Tysoe C, et al. A new type of pharmacological chaperone for GM1-gangliosidosis related human lysosomal β-galactosidase: N-Substituted 5-amino-1-hydroxymethyl-cyclopen-tanetriols. Bioorg Med Chem Lett 2017; 27(15): 3431-5.
[http://dx.doi.org/10.1016/j.bmcl.2017.05.086] [PMID: 28600 215] ]. In the absence of a specific treatment, especially in Morocco, our patient was initially given symptomatic treatment: Treatment of pulmonary and urinary infections, motor and respiratory physiotherapy. The infant died as a result of severe neurological deterioration before a genetic study could be performed.
CONCLUSION
The broad variability of clinical symptoms in patients with GM1-gangliosidosis disease may cause delays in diagnosis.
In the absence of screening programmes, the finding of persistent, extensive slate-grey Mongolian spots in newborns or infants could lead to early detection and treatment of GM1-gangliosidosis before irreversible organ damage occurs.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this review.
CONSENT FOR PUBLICATION
A written informed consent was obtained from the parents when they were enrolled.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors thank the patient's parents, Dr. Klevor Raymond, and the staff of the pediatric department for their help and support.
REFERENCES
| [1] | Ida H, Eto Y, Maekawa K. Fetal GM1-gangliosidosis: Morphological and biochemical studies. Brain Dev 1989; 11(): 394-8. [http://dx.doi.org/10.1016/S0387-7604(89)80023-3] |
| [2] | Lee JS, Choi JM, Lee M, et al. Diagnostic challenge for the rare lysosomal storage disease: Late infantile GM1-gangliosidosis. Brain Dev 2018; 40(5): 383-90. [http://dx.doi.org/10.1016/j.braindev.2018.01.009] [PMID: 294 39846] |
| [3] | Abilkassem R, Agadr A. Tache mongoloïde extensive: Un signe clinique qui mérite une attention particulière. Pan Afr Med J 2013; 16: 41. [http://dx.doi.org/10.11604/pamj.2013.16.41.3062] [PMID: 246 48854] |
| [4] | Sperb F, Vairo F, Burin M, Mayer FQ, Matte U, Giugliani R. Genotypic and phenotypic characterization of Brazilian patients with GM1-gangliosidosis. Gene 2013; 512(1): 113-6. [http://dx.doi.org/10.1016/j.gene.2012.09.106] [PMID: 23046 582] |
| [5] | Yang CF, Wu JY, Tsai FJ. Three novel beta-galactosidase gene mutations in Han Chinese patients with GM1-gangliosidosis are correlated with disease severity. J Biomed Sci 2010; 17: 79. [http://dx.doi.org/10.1186/1423-0127-17-79] [PMID: 209202 81] |
| [6] | Armstrong-Javors A, Chu CJ. Child neurology: Exaggerated dermal melanocytosis in a hypotonic infant: A harbinger of GM1-gangliosidosis. Neurology 2014; 83(17): e166-8. [http://dx.doi.org/10.1212/WNL.0000000000000912] [PMID: 253324 52] |
| [7] | Caciotti A, Garman SC, Rivera-Colón Y, et al. GM1-gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochim Biophys Acta 2011; 1812(7): 782-90. [http://dx.doi.org/10.1016/j.bbadis.2011.03.018] [PMID: 21497194] |
| [8] | Ashrafi MR, Shabanian R, Mohammadi M, Kavusi S. Extensive Mongolian spots: A clinical sign merits special attention. Pediatr Neurol 2006; 34(2): 143-5. [http://dx.doi.org/10.1016/j.pediatrneurol.2005.07.010] [PMID: 1645882 9] |
| [9] | Kumar Bhardwaj N, Khera D. Mongolian spots in GM1-Gangliosidosis. Indian Pediatr 2016; 53(12): 1133. [PMID: 28064276] |
| [10] | Mimouni-Bloch A, Finezilber Y, Rothschild M, Raas-Rothschild A. Extensive Mongolian Spots and Lysosomal Storage Diseases. J Pediatr 2016; 170: 333-e1. [http://dx.doi.org/10.1016/j.jpeds.2015.11.009] [PMID: 267038 76] |
| [11] | Hackbart BA, Arita JH, Pinho RS, Masruha MR, Vilanova LCP. Mongolian spots are not always a benign sign. J Pediatr 2013; 162(5): 1070. [http://dx.doi.org/10.1016/j.jpeds.2012.12.044] [PMID: 23374 678] |
| [12] | Weissbluth M, Esterly NB, Caro WA. Report of an infant with GM1-gangliosidosis type I and extensive and unusual mongolian spots. Br J Dermatol 1981; 104(2): 195-200. [http://dx.doi.org/10.1111/j.1365-2133.1981.tb00045.x] [PMID: 678306 1] |
| [13] | Jarnes Utz JR, Kim S, King K, et al. Infantile gangliosidoses: Mapping a timeline of clinical changes. Mol Genet Metab 2017; 121(2): 170-9. [http://dx.doi.org/10.1016/j.ymgme.2017.04.011] [PMID: 2847 6546] |
| [14] | Stoica L, Todeasa S, Lata J, Martin D, Seana-Esteves M. Optimization of AAV-gene therapy for GM1-gangliosidosis. Mol Ther 2016; 24: 149. [http://dx.doi.org/10.1016/S1525-0016(16)33182-3] |
| [15] | Yuki M, Keiichi M, Taishi H, et al. Feasibility study of cyclodextrins as active pharmaceutical ingredients for the treatment of GM1-gangliosidosis. Asian journal of pharmaceutical sciences 2016; 11: 183-4. |
| [16] | Schalli M, Weber P, Tysoe C, et al. A new type of pharmacological chaperone for GM1-gangliosidosis related human lysosomal β-galactosidase: N-Substituted 5-amino-1-hydroxymethyl-cyclopen-tanetriols. Bioorg Med Chem Lett 2017; 27(15): 3431-5. [http://dx.doi.org/10.1016/j.bmcl.2017.05.086] [PMID: 28600 215] |