- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




The Open Conference Proceedings Journal
(Biological Sciences, Chemical Sciences, Physical Sciences, Medicine, Engineering & Technology)
(Discontinued)
ISSN: 2210-2892 ― Volume 10, 2020
Constraints of Drug Resistance in Mycobacterium tuberculosis - Prospects for Pharmacological Reversion of Susceptibility to Antibiotics
Aleksandr I. Ilin*, 1, Murat E. Kulmanov1, Ilya S. Korotetskiy1, Marina V. Lankina1, Gulshara K. Akhmetova1, Sergey V. Shvidko1, Oleg N. Reva*, 2
Abstract
Emergence of multidrug resistant strains of Mycobacterium tuberculosis (MDR-TB) threatens humanity. This problem was complicated by the crisis in development of new anti-tuberculosis antibiotics. Induced reversion of drug resistance seems promising to overcome the problem. Successful clinical trial of a new anti-tuberculosis nanomolecular complex FS-1 has demonstrated prospectively of this approach in combating MDR-TB. Several clinical MDR-TB cultures were isolated from sputum samples prior and in the process of the clinical trial. Every isolate was tested for susceptibility to antibiotics and then they were sequenced for comparative genomics. It was found that the treatment with FS-1 caused an increase in the number of antibiotic susceptible strains among Mtb isolates that was associated with a general increase of genetic heterogeneity of the isolates. Observed impairing of phthiocerol dimycocerosate biosynthesis by disruptive mutations in ppsACD subunits indicated a possible virulence remission for the sake of persistence. It was hypothesized that the FS-1 treatment eradicated the most drug resistant Mtb variants from the population by aggravating the fitness cost of drug resistance mutations. Analysis of distribution of these mutations in the global Mtb population revealed that many of them were incompatible with each other and dependent on allelic states of many other polymorphic loci. The latter discovery may explain the negative correlation between the genetic heterogeneity of the population and the level of drug tolerance. To the best of our knowledge, this work was the first experimental confirmation of the drug induced antibiotic resistance reversion by the induced synergy mechanism that previously was predicted theoretically.
Article Information

Identifiers and Pagination:
Year: 2017Volume: 8
First Page: 33
Last Page: 43
Publisher Id: TOPROCJ-8-33
DOI: 10.2174/2210289201708010033
Article History:
Received Date: 26/08/2016Revision Received Date: 11/12/2016
Acceptance Date: 03/01/2017
Electronic publication date: 22/03/2017
Collection year: 2017
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: https://creativecommons.org/licenses/by/4.0/legalcode. This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
* Address correspondence to these authors at the Centre for Bioinformatics and Computational Biology, Department of Biochemistry, University of Pretoria, Hillcrest, Lynnwood Rd, Pretoria 0002, South Africa; Tel: +27-12-420-5810; Fax: +27-12-420-5800; E-mail: oleg.reva@up.ac.za;
Scientific Center for Anti-Infectious Drugs (SCAID), 75V Al-Farabi Ave, Almaty 050060, Kazakhstan; Tel: +8-727-266-4832; E-mail: ilin_ai@mail.ru
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 26-08-2016 |
Original Manuscript | Constraints of Drug Resistance in Mycobacterium tuberculosis - Prospects for Pharmacological Reversion of Susceptibility to Antibiotics | |
1. INTRODUCTION
Public health is threatened by drug resistant infection agents. Many contagious diseases from the past have spread around the world exacerbating measures for epidemiological control around the world. Until now there is no confidence among scientists by which means the problem may be taken under control [1Tenover, F.C. Development and spread of bacterial resistance to antimicrobial agents: an overview. Clin. Infect. Dis., 2001, 33(Suppl. 3), S108-S115.
[http://dx.doi.org/10.1086/321834] [PMID: 11524705] , 2Morens, D.M.; Folkers, G.K.; Fauci, A.S. The challenge of emerging and re-emerging infectious diseases. Nature, 2004, 430(6996), 242-249.
[http://dx.doi.org/10.1038/nature02759] [PMID: 15241422] ]. Tackling of the drug resistant infections requires coordinated international actions, development of new antibiotics and introducing of innovative approaches of drug induced reversion of susceptibility of pathogens to conventional antibiotics [3Baym, M.; Stone, L.K.; Kishony, R. Multidrug evolutionary strategies to reverse antibiotic resistance. Science, 2016, 351(6268), aad3292.
[http://dx.doi.org/10.1126/science.aad3292] [PMID: 26722002] , 4O’Neill, J. Tackling drug resistance globally: final report and recommendations. The Review on Antimicrobial Resistance, 2016. Available at: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf].
Mycobacterium tuberculosis (Mtb) is the causative agent of tuberculosis, which accounts for a significant death toll around the world. Ninety percent of tuberculosis cases occur in developing countries due to inadequacies in the healthcare resources and patient follow-up [5Nouvel, L.X.; Kassa-Kelembho, E.; Dos Vultos, T.; Zandanga, G.; Rauzier, J.; Lafoz, C.; Martin, C.; Blazquez, J.; Talarmin, A.; Gicquel, B. Multidrug-resistant Mycobacterium tuberculosis, Bangui, Central African Republic. Emerg. Infect. Dis., 2006, 12(9), 1454-1456.
[http://dx.doi.org/10.3201/eid1209.060361] [PMID: 17073103] ]. The problem became global with the emergence of multidrug resistant strains of Mtb (MDR-TB), which currently are reported in all countries with the burden of disease not witnessed since before the advent of antibiotics [6Cohn, D.L.; Bustreo, F.; Raviglione, M.C. Drug-resistant tuberculosis: review of the worldwide situation and the WHO/IUATLD Global Surveillance Project. Clin Infect Dis., 1997, 24(Suppl. 1), 7Zager, E.M.; McNerney, R. Multidrug-resistant tuberculosis. BMC Infect. Dis., 2008, 8, 10.
[http://dx.doi.org/10.1186/1471-2334-8-10] [PMID: 18221534] ].
Mtb is known as a highly clonal bacterium meaning that it is generally resistant to horizontal gene transfer. All cases of acquired resistance to anti-tuberculosis drugs resulted from spontaneous mutations in genes encoding functional proteins or in promoter regions of these genes [7Zager, E.M.; McNerney, R. Multidrug-resistant tuberculosis. BMC Infect. Dis., 2008, 8, 10.
[http://dx.doi.org/10.1186/1471-2334-8-10] [PMID: 18221534] ]. As per today, 1,031 mutations were discovered in Mtb rendering resistance to nine major groups of anti-tuberculosis drugs – aminoglycosides, ethambutol, ethionamide, fluoroquinolones, isoniazid, para-aminosalisylic acid, pyrazinamide, rifampicin and streptomycin [8Sandgren, A.; Strong, M.; Muthukrishnan, P.; Weiner, B.K.; Church, G.M.; Murray, M.B. Tuberculosis drug resistance mutation database. PLoS Med., 2009, 6(2), e2.
[http://dx.doi.org/10.1371/journal.pmed.1000002] [PMID: 19209951] ]. Multidrug resistance is rendered by combinations of these mutations. To overcome MDR, new antibiotics are in great demand, which preferably should be chemically unrelated to the conventional antibiotics and should be aimed at alternative molecular targets to succeed with curation of MDR-TB.
Progress in design of new anti-TB drugs over the last decades was inappropriate to the challenge of global distribution of the MDR-TB infection [9Van, den Boogaard, J.; Kibiki, G.S.; Kisanga, E.R. New drugs against tuberculosis: problems, progress, and evaluation of agents in clinical development. Antimicrob Agents Chemother, 2009, 53(3), 849-862.]. Only a few anti-TB drugs have been developed recently including Bedaquiline [10Mahajan, R. Bedaquiline: First FDA-approved tuberculosis drug in 40 years. Int. J. Appl. Basic Med. Res., 2013, 3(1), 1-2.
[http://dx.doi.org/10.4103/2229-516X.112228] [PMID: 23776831] ], Delamanid [11Gupta, R.; Geiter, L.J.; Wells, C.D.; Gao, M.; Cirule, A.; Xiao, H. Delamanid for extensively drug-resistant tuberculosis. N. Engl. J. Med., 2015, 373(3), 291-292.
[http://dx.doi.org/10.1056/NEJMc1415332] [PMID: 26176402] ] and FS-1 [12Ilin, A.I.; Kulmanov, M.E. Antibacterial agent for treating infectious diseases of bacterial origin. Patent WO 2012091534 A1, 2012.] after a long period of almost 40 years when no new anti-TB drugs had been registered. FS-1 was of special interest for this study because of a reported induction of the drug resistance reversion in MDR-TB. This phenomenon was observed during the clinical trial and in series of in vitro experiments. The idea of reversion of susceptibility of pathogens to conventional antibiotics by supplementing them with some supporting drugs was suggested several decades ago [13Jagannath, C.; Reddy, V.M.; Gangadharam, P.R. Enhancement of drug susceptibility of multi-drug resistant strains of Mycobacterium tuberculosis by ethambutol and dimethyl sulphoxide. J. Antimicrob. Chemother., 1995, 35(3), 381-390.
[http://dx.doi.org/10.1093/jac/35.3.381] [PMID: 7782254] ]. Currently, this approach gained a general recognition because of the crises in development of new antibiotics. Several theoretical models of antibiotic resistance reversion were discussed in a recently published review [3Baym, M.; Stone, L.K.; Kishony, R. Multidrug evolutionary strategies to reverse antibiotic resistance. Science, 2016, 351(6268), aad3292.
[http://dx.doi.org/10.1126/science.aad3292] [PMID: 26722002] ]. However, this review lacked solid statistical and experimental grounding for these hypotheses. It should be accepted that the absence of experimental models of drug induced antibiotic resistance reversion was the major impediment in this field of research. Induction of drug resistance reversion in MDR-TB by FS-1 made it possible to study this phenomenon on the genetic level. Furthermore, statistical analysis of distribution of polymorphic sites in the global Mtb population based on the data from GMTV database [14Chernyaeva, E.N.; Shulgina, M.V.; Rotkevich, M.S.; Dobrynin, P.V.; Simonov, S.A.; Shitikov, E.A.; Ischenko, D.S.; Karpova, I.Y.; Kostryukova, E.S.; Ilina, E.N.; Govorun, V.M.; Zhuravlev, V.Y.; Manicheva, O.A.; Yablonsky, P.K.; Isaeva, Y.D.; Nosova, E.Y.; Mokrousov, I.V.; Vyazovaya, A.A.; Narvskaya, O.V.; Lapidus, A.L.; OBrien, S.J. Genome-wide Mycobacterium tuberculosis variation (GMTV) database: a new tool for integrating sequence variations and epidemiology. BMC Genomics, 2014, 15, 308.
[http://dx.doi.org/10.1186/1471-2164-15-308] [PMID: 24767249] ] allowed getting insight into functional relations between drug resistance mutations of Mtb. A work hypothesis was that FS-1 could cause an active counter-selection of drug resistant variants from Mtb populations by aggravating the cumulated fitness cost of the drug resistance mutations as it was predicted previously by a mathematical model by Cohen and Murray [15Cohen, T.; Murray, M. Modeling epidemics of multidrug-resistant M. tuberculosis of heterogeneous fitness. Nat. Med., 2004, 10(10), 1117-1121.
[http://dx.doi.org/10.1038/nm1110] [PMID: 15378056] ]. The fitness cost reduces viability and competitiveness of the strains bearing drug resistance mutations [16Cohen, T.; Sommers, B.; Murray, M. The effect of drug resistance on the fitness of Mycobacterium tuberculosis. Lancet Infect. Dis., 2003, 3(1), 13-21.
[http://dx.doi.org/10.1016/S1473-3099(03)00483-3] [PMID: 12505028] ]. However, consequent compensatory mutations reduce the fitness cost to the level of the initial drug susceptible strains [17Pym, A.S.; Saint-Joanis, B.; Cole, S.T. Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect. Immun., 2002, 70(9), 4955-4960.
[http://dx.doi.org/10.1128/IAI.70.9.4955-4960.2002] [PMID: 12183541] -19Luciani, F.; Sisson, S.A.; Jiang, H.; Francis, A.R.; Tanaka, M.M. The epidemiological fitness cost of drug resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA, 2009, 106(34), 14711-14715.
[http://dx.doi.org/10.1073/pnas.0902437106] [PMID: 19706556] ] facilitating wide distribution of the MDR-TB infection. Nevertheless, overcoming drug resistance by aggravating the fitness cost seems promising. Our results demonstrated that several potential drug resistance mutations were removed from the population during the therapeutic treatment with conventional antibiotics supplemented with FS-1. This treatment also caused an accumulation of disruptive mutations in ppsACD and numerous random mutations that led to weakening of the drug resistance and possibly virulence of the pathogens compared to the initial isolates obtained before the beginning of the treatment. This correlation may be explained in a way that the random mutations could impair the general genetic context necessitated for an effective drug resistance phenotype.
2. METHODS
2.1. Clinical Trial of FS-1 and Mtb Isolation
Second phase of the clinical trial of FS-1 against MDR-TB was carried out in Kazakhstan in 2015. Mtb strains have been isolated on a regular basis from patients’ sputum samples before the treatment beginning and during the therapy with FS-1 supplementing the standard regiment of antibiotic treatment of MDR-TB. The complex of anti-tuberculosis antibiotics included pyrazinamide (per os 40 mg/kg), cycloserine (per os 20.0 mg/kg), prothionamide (per os 20.0 mg/kg), capreomycin (intramuscular 20.0 mg/kg) and amikacin (intramuscular 28.0 mg/kg). FS-1 was administrated per os in 2.5 mg/kg in 30 min prior to antibiotics. Sputum samples were inoculated into liquid Löwenstein-Jensen medium (HiMedia Laboratories, India) and cultivated at 37°C for 8 weeks. Culture growth was controlled visually, by microscopy of Ziehl–Neelsen stained smears and by standard diagnostic biochemical tests including positive catalase, niacin and nicotinamidase activities, negative Tween-80 hydrolysis and susceptibility to sodium salicylate [20Bloch, H.; Segal, W. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J. Bacteriol., 1956, 72(2), 132-141.
[PMID: 13366889] ].
2.2. Susceptibility to Antibiotics
Susceptibility to antibiotics was tested by growing the cultures for 4 weeks at 37°C in test-tubes on solid Löwenstein-Jensen medium (HiMedia Laboratories, India; pH 6.8) supplemented with antibiotics in concentrations recommended by WHO [21World Health Organization. Interim Policy Guidance on Drug Susceptibility Testing (DST) of Second-Line Anti-Tuberculosis Drugs World Health Organization Document, 2008. WHO/HTM/TB/2008.392] and in the literature [22Krüüner, A.; Yates, M.D.; Drobniewski, F.A. Evaluation of MGIT 960-based antimicrobial testing and determination of critical concentrations of first- and second-line antimicrobial drugs with drug-resistant clinical strains of Mycobacterium tuberculosis. J. Clin. Microbiol., 2006, 44(3), 811-818.
[http://dx.doi.org/10.1128/JCM.44.3.811-818.2006] [PMID: 16517859] ]. The following antibiotics were used: isoniazid (0.2 μg/ml), rifampicin (40.0 μg/ml), streptomycin (4.0 μg/ml), ethambutol (2.0 μg/ml), amikacin (30.0 μg/ml), kanamycin (30.0 μg/ml), capreomycin (40.0 μg/ml), ofloxacin (2.0 μg/ml), cycloserine (40.0 μg/ml), ethionamide (40.0 μg/ml) and pyrazinamide (200.0 μg/ml on FAST-3L medium, BIOK, Russia; pH 5.0).
2.3. Sequencing of Mtb Isolates and Variant Calling
DNA samples were extracted from the Mtb isolates by the Cetyltrimethylammonium bromide (CTAB) method [23Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res., 1980, 8(19), 4321-4325.
[http://dx.doi.org/10.1093/nar/8.19.4321] [PMID: 7433111] ]. The eluted DNA was quantified using the Qubit dsDNA BR Assay Kit (Life Technologies, USA). The samples were sequenced by Macrogen (South Korea) using the Illumina HiSeq 2000 paired-end sequencing technology. Produced read coverage was in the range of 800-1000. Raw DNA reads were deposited at EMBL-ENA under the reference numbers ERR1559736-43. The genome sequence of three clinical MDR-TB isolates M. tuberculosis SCAID 187.0 [24Ilin, A.I.; Kulmanov, M.E.; Korotetskiy, I.S.; Akhmetova, G.K.; Lankina, M.V.; Shvidko, S.V.; Reva, O.N. Complete genome sequence of multi-drug resistant clinical isolate Mycobacterium tuberculosis 187.0 used to study an effect of drug susceptibility reversion by a new medicinal drug FS-1. Genome Announc., 2015, 3(6), e01272-e15.
[http://dx.doi.org/10.1128/genomeA.01272-15] [PMID: 26543112] ], 252.0 and 320.0 were deposited in NCBI with accession numbers CP012506, CP016888 and CP016794, respectively. CLC Genomics Workbench 7.0.3 was used for sequence assembly and variant calling by the quality-based variant detection algorithm.
2.4. Statistical Analysis of Mtb Genomic Polymorphism
In the current work, 58,025 amino acid substitutions in 1,623 Mtb strains recorded in GMTV database [14Chernyaeva, E.N.; Shulgina, M.V.; Rotkevich, M.S.; Dobrynin, P.V.; Simonov, S.A.; Shitikov, E.A.; Ischenko, D.S.; Karpova, I.Y.; Kostryukova, E.S.; Ilina, E.N.; Govorun, V.M.; Zhuravlev, V.Y.; Manicheva, O.A.; Yablonsky, P.K.; Isaeva, Y.D.; Nosova, E.Y.; Mokrousov, I.V.; Vyazovaya, A.A.; Narvskaya, O.V.; Lapidus, A.L.; OBrien, S.J. Genome-wide Mycobacterium tuberculosis variation (GMTV) database: a new tool for integrating sequence variations and epidemiology. BMC Genomics, 2014, 15, 308.
[http://dx.doi.org/10.1186/1471-2164-15-308] [PMID: 24767249] ] were analyzed for their distribution in the global Mtb population. Information about drug associated mutations was obtained from the TB Drug Resistance Mutation Database [8Sandgren, A.; Strong, M.; Muthukrishnan, P.; Weiner, B.K.; Church, G.M.; Murray, M.B. Tuberculosis drug resistance mutation database. PLoS Med., 2009, 6(2), e2.
[http://dx.doi.org/10.1371/journal.pmed.1000002] [PMID: 19209951] ].
For every pair of SNP the multi-allelic linkage disequilibrium LD (1) and R2 (2) were calculated [25Yang, J.; Shikano, T.; Li, M-H.; Merilä, J. Genome-wide linkage disequilibrium in nine-spined stickleback populations. G3 (Bethesda), 2014, 4(10), 1919-1929.
[http://dx.doi.org/10.1534/g3.114.013334] [PMID: 25122668] ]:
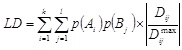 |
(1) |
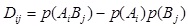 |
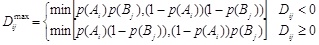 |
where k and l are the numbers of alleles for loci A and B,
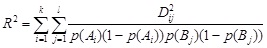 |
(2) |
Graphviz 2.24 and in-house Python scripts were used for grouping and visualization of functionally linked alleles. Complete genome sequences were aligned by Mauve [26Darling, A.C. Mau. B.; Blattner, F.D.; Perna, N.T. Mauve: multiple alignment of conserved genomic sequences with rearrangements. Genet. Res., 2004, 14(7), 1394-1403.
[http://dx.doi.org/10.1101/gr.2289704] ].
3. RESULTS AND DISCUSSION
3.1. Analysis of Distribution of Drug Resistance Mutations in the Global Mtb Population
Linkage disequilibrium (LD) indicates the level of dependence between allelic states in different polymorphic loci on genomes. LD varies from 1 (non-random association of two mutations) to -1 (displacement of one mutation by another due to a functional or evolutionary incompatibility). Strong LD may be explained by a functional interplay between two genes, but also it may result from a functionally neutral inheritance of independent mutations from one common ancestor known as the genetic drift.
Many drug resistance mutations showed a strong negative LD in relation to each other. In Fig. (1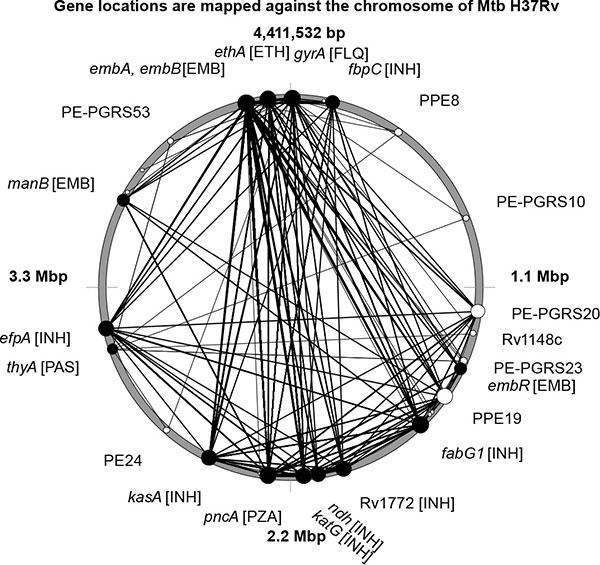 ), several drug resistance mutations with LD ≤ -0.9 were plotted on the circular chromosome of the Mtb type strain H37Rv. The most likely explanation for the negative LD is that the cumulated fitness cost associated with these mutations drastically decreased viability of the mutant strains. Top 20 drug resistance mutations showing significant incompatibility with other mutations are shown in Table 1.
), several drug resistance mutations with LD ≤ -0.9 were plotted on the circular chromosome of the Mtb type strain H37Rv. The most likely explanation for the negative LD is that the cumulated fitness cost associated with these mutations drastically decreased viability of the mutant strains. Top 20 drug resistance mutations showing significant incompatibility with other mutations are shown in Table 1.

The top 20 polymorphic alleles negatively displacing other drug resistance mutations were localized in six genes encoding embAB arabinosyl-indolyl-acetylinositol synthase, pncA pyrazinamidase-nicotinamidase, fabG1 3-oxoacyl-[acyl]-carrier reductase, kasA 3-oxoacyl-[acyl]-carrier protein synthase, ethA monooxygenase, katG catalase-peroxidase-peroxynitritase and ndh NADH dehydrogenase. They showed strong negative LD links with other drug resistance mutations in inhA NADH-dependent enoyl-[acyl]-carrier-protein reductase; fbpC mycolyl transferase; fabD malonyl CoA-acyl carrier protein transacylase; efpA integral membrane efflux pump; Rv1772 and Rv2242 hypothetical proteins (isoniazid resistance); manB D-alpha-D-mannose-1-phosphate guanylyltransferase; rmlD dTDP-6-deoxy-L-lyxo-4-hexulose reductase, embC membrane indolylacetylinositol arabinosyltransferase and embR transcriptional regulator (ethambutol resistance); gyrA DNA topoisomerase subunit (resistance to fluoroquinolones); thyA thymidylate synthase (para-aminosalisylic acid resistance); rpsL 30S ribosomal protein and gid glucose-inhibited division protein (streptomycin resistance).
Positive LD may indicate synergetic relations between drug resistance mutations leading to an increased tolerance to one or multiple antibiotics. In general, the positive links between drug resistance mutations were not as strong as the negative displacements. Loci linked by LD ≥ 0.75 are shown in Fig. (2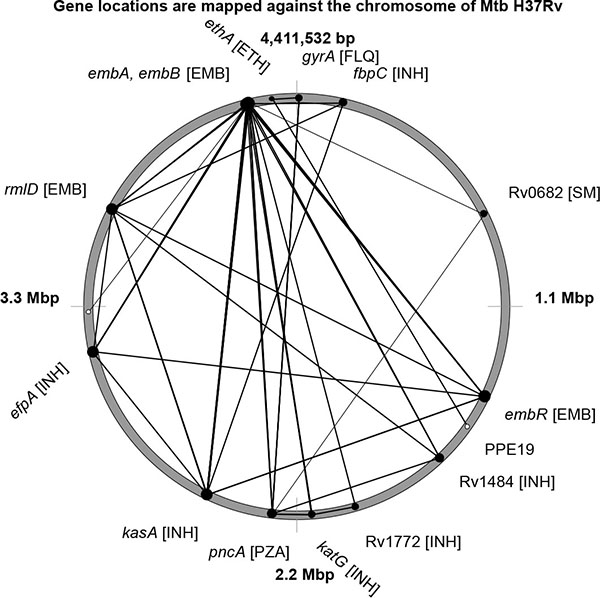 ).
).
 |
Fig. (2) Location of drug resistance mutations on the chromosome of Mtb H37Rv are depicted by black cycles. Pairs of alleles showing strong positive LD are linked by lines. |
Mutations in four genes: embB, embC, embR and rmlD, all rendering the ethambutol resistance, showed strong positive LD links. These mutations formed the basis of multidrug resistance in Mtb due to their compatibility with many other drug resistance mutations. Other patterns of multidrug resistance were grouped around mutations in katG, kasA, fbpC and efpA.
Relations between drug resistance mutations and other polymorphic alleles, which could be possible compensatory mutations, were identified by R2 linkage coefficients. The biggest cluster of co-evolved polymorphic alleles in Fig. (3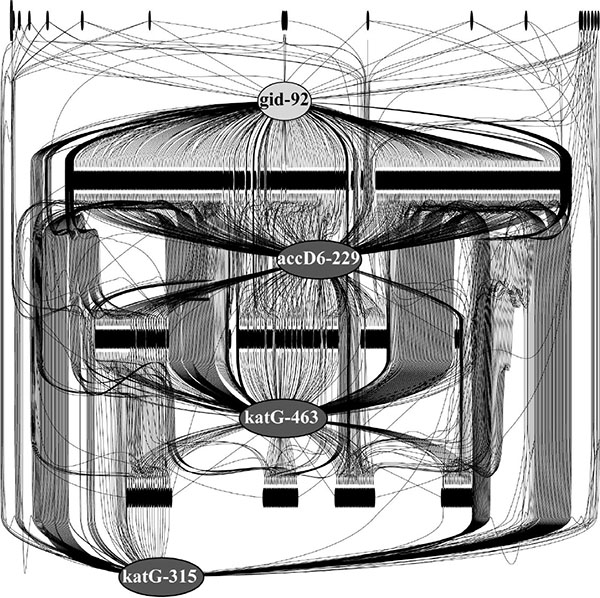 ) is centered round four drug resistance mutations in the 92nd codon of gid (streptomycin resistance) and in the 229th codon of accD6, 463rd codon and 315th codon of katG, all of them rendering the isoniazid resistance.
) is centered round four drug resistance mutations in the 92nd codon of gid (streptomycin resistance) and in the 229th codon of accD6, 463rd codon and 315th codon of katG, all of them rendering the isoniazid resistance.
Black bars in Fig. (3) depict numerous polymorphic alleles in Mtb genomes showing positive correlations with the drug resistance mutations. Despite they not have been reported in the literature as drug resistance mutations, the strong R2 relations of these polymorphisms with the canonical antibiotic resistance sites suggested that at least some of them may be of importance for rendering the drug resistance in Mtb and/or reducing the associated fitness costs.
This analysis showed that out of more than 1,000 known drug resistance mutations only a limited number of them are compatible with each other and even the compatible mutations depend on the allelic states of polymorphisms in many other genes.
3.2. Genetic Insight into Antibiotic Resistance Reversion Induced by FS-1
In total, 236 MDR-TB isolates were obtained during the clinical trial of FS-1. Mtb cultures were isolated prior to the treatment and then on a weekly basis during the combined treatment course by the antibiotics and FS-1. A gradual reversion of drug resistance to a more sensitive phenotype was observed in series of isolates despite the selective pressure of antibiotics (Table 2). The treatment with FS-1 caused the reversion of drug resistance to aminoglycoside antibiotics (kanamycin, amikacin and capreomycin); ethambutol and cycloserine that was not the case in the control group of patients treated by the standard regiment. Selected Mtb isolates were sequenced to study genomic changes, which could explain the observed drug resistance reversion (Table 2). Complete genome sequences of the initial Mtb isolates from the patients #187, #252 and #320 were deposited in NCBI with the respective accession numbers CP012506, CP016888 and CP016794. Then these sequences were used as references for variant calling. Comparative analysis of the sequenced genomes (marked SCAID #) by Mauve alignment against several known Mtb genomes from NCBI revealed their belonging to the Beijing clade (Fig. 4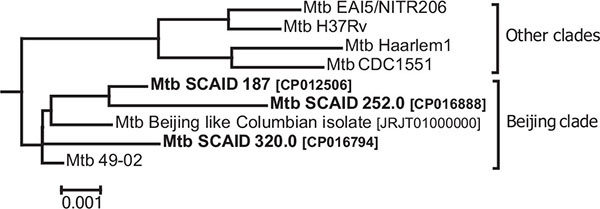 ).
).

 |
Fig. (4) Phylogenetic relations between the clinical SCAID isolates used in this study and several selected reference Mtb strains. |

Beijing clade is characterized by an innate antibiotic resistance [27Merker, M.; Blin, C.; Mona, S.; Duforet-Frebourg, N.; Lecher, S.; Willery, E.; Blum, M.G.; Rüsch-Gerdes, S.; Mokrousov, I.; Aleksic, E.; Allix-Béguec, C.; Antierens, A.; Augustynowicz-Kopeć, E.; Ballif, M.; Barletta, F.; Beck, H.P.; Barry, C.E., III; Bonnet, M.; Borroni, E.; Campos-Herrero, I.; Cirillo, D.; Cox, H.; Crowe, S.; Crudu, V.; Diel, R.; Drobniewski, F.; Fauville-Dufaux, M.; Gagneux, S.; Ghebremichael, S.; Hanekom, M.; Hoffner, S.; Jiao, W.W.; Kalon, S.; Kohl, T.A.; Kontsevaya, I.; Lillebæk, T.; Maeda, S.; Nikolayevskyy, V.; Rasmussen, M.; Rastogi, N.; Samper, S.; Sanchez-Padilla, E.; Savic, B.; Shamputa, I.C.; Shen, A.; Sng, L.H.; Stakenas, P.; Toit, K.; Varaine, F.; Vukovic, D.; Wahl, C.; Warren, R.; Supply, P.; Niemann, S.; Wirth, T. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat. Genet., 2015, 47(3), 242-249.
[http://dx.doi.org/10.1038/ng.3195] [PMID: 25599400] ]. Eight canonical drug resistance mutations were found in the three sequenced Mtb strains [8Sandgren, A.; Strong, M.; Muthukrishnan, P.; Weiner, B.K.; Church, G.M.; Murray, M.B. Tuberculosis drug resistance mutation database. PLoS Med., 2009, 6(2), e2.
[http://dx.doi.org/10.1371/journal.pmed.1000002] [PMID: 19209951] ]: GyrAAsp:94→Asn/Gly and GyrASer:95→Thr (FLQ); RpsLLys:43→Arg (SM); KatGSer:315→Thr and KatGArg:463→Leu (INH); AccD6Gln:141→Pro (PZA); PncAAsp:229→Gly (INH); EmbBMet:306→Ile (EMB); and gidGlu:92→Asp (SM). Despite of the classical drug resistance mutations in KatG, all three strains were catalase positive. All these strains were resistant to ethionamide (Table 2). In Mtb this resistance usually is conferred by mutations in ethA [8Sandgren, A.; Strong, M.; Muthukrishnan, P.; Weiner, B.K.; Church, G.M.; Murray, M.B. Tuberculosis drug resistance mutation database. PLoS Med., 2009, 6(2), e2.
[http://dx.doi.org/10.1371/journal.pmed.1000002] [PMID: 19209951] ]. In the strain SCAID 320.0 this gene was truncated in the mid by a frame shift mutation that probably made this gene completely nonfunctional in this strain. The strain SCAID 187.0 has a Leu:82→Pro substitution in ethA. It was not reported as an ETH resistance mutation, but it is located very close to other known mutations of this drug resistance. The strain SCAID 252.0 possesses an Ala:248→Asp substitution in the area of other drug resistance mutations within this gene; however, rendering ethionamide resistance by this mutation has not been reported either. Another mutation EthALeu:190→Phe became abundant in the population SCAID 252 on the 9th week of the treatment with antibiotics and FS-1, but it disappeared in the later isolates. Canonical mutations in ethA are incompatible with many other drug resistance mutations (Fig. 1) that may explain their absence in these MDR-TB strains. Therapy with FS-1 prevented the accumulation of new potential drug resistance mutations in the SCAID 252 isolates. Accumulation of other mutations, which potentially could lead to drug resistance, was observed also in the isolate SCAID 187.9. Substitutions Phe:95→Leu in cyclohexanone monooxygenase Rv0565c and Arg:264→Gly in CydC cytochrome component transporter were found in 60% of corresponding DNA reads. Both these genes belong to potential prodrug activators [28Fraaije, M.W.; Kamerbeek, N.M.; Heidekamp, A.J.; Fortin, R.; Janssen, D.B. The prodrug activator EtaA from Mycobacterium tuberculosis is a Baeyer-Villiger monooxygenase. J. Biol. Chem., 2004, 279(5), 3354-3360.
[http://dx.doi.org/10.1074/jbc.M307770200] [PMID: 14610090] ]; however, no drug resistance mutations have been reported in these genes in the literature until now. No additional drug resistance mutations were observed in the SCAID 320 isolates.
An increased frequency of mutations was observed in subunits ppsACD of phenolphthiocerol polyketide synthase in all series of isolates. Frame shift indels were found in the genes ppsC and ppsD in the isolates 187.7 and 187.9 with the frequencies up to 60-100%. Substitutions His→Pro in proline rich linker regions between the acyltransferase and dehydratase domains were observed in the isolates 252.9 (30% of reads only in ppsC) and 252.12 (up to 60% of reads in ppsA and ppsC). In the isolate 320.8, this mutation was found in 50% of reads aligned against ppsA. The initial cultures 187.0, 252.0 and 320.0 were free from these mutations. Phthiocerol dimycocerosate is a characteristic cell wall compound of Mycobacteria and an important virulence factor [29Yu, J.; Tran, V.; Li, M.; Huang, X.; Niu, C.; Wang, D.; Zhu, J.; Wang, J.; Gao, Q.; Liu, J. Both phthiocerol dimycocerosates and phenolic glycolipids are required for virulence of Mycobacterium marinum. Infect. Immun., 2012, 80(4), 1381-1389.
[http://dx.doi.org/10.1128/IAI.06370-11] [PMID: 22290144] ]. However, it was recently published that impaired phthiocerol biosynthesis was associated with an enhanced ability to persist in host organisms [30Torrey, H.L.; Keren, I.; Via, L.E.; Lee, J.S.; Lewis, K. High persister mutants in Mycobacterium tuberculosis. PLoS One, 2016, 11(5) e0155127.
[http://dx.doi.org/10.1371/journal.pone.0155127] [PMID: 27176494] ].
Proline rich linkers are important for an appropriate spatial structuring of multidomain proteins [31Williamson, M.P.; Keren, I.; Via, L.E. The structure and function of proline-rich regions in proteins. Biochem. J., 1994, 297(Pt 2), 249-260.
[http://dx.doi.org/10.1042/bj2970249] [PMID: 8297327] ]. Introducing of extra proline residues may alter the efficacy of the phthiocerol biosynthesis in the same way as shift mutations; however, it has not been proved experimentally for these genes yet.
Another commonality of the studied series of isolates was an increase of genetic heterogeneity of the populations during the treatment course (Fig. 5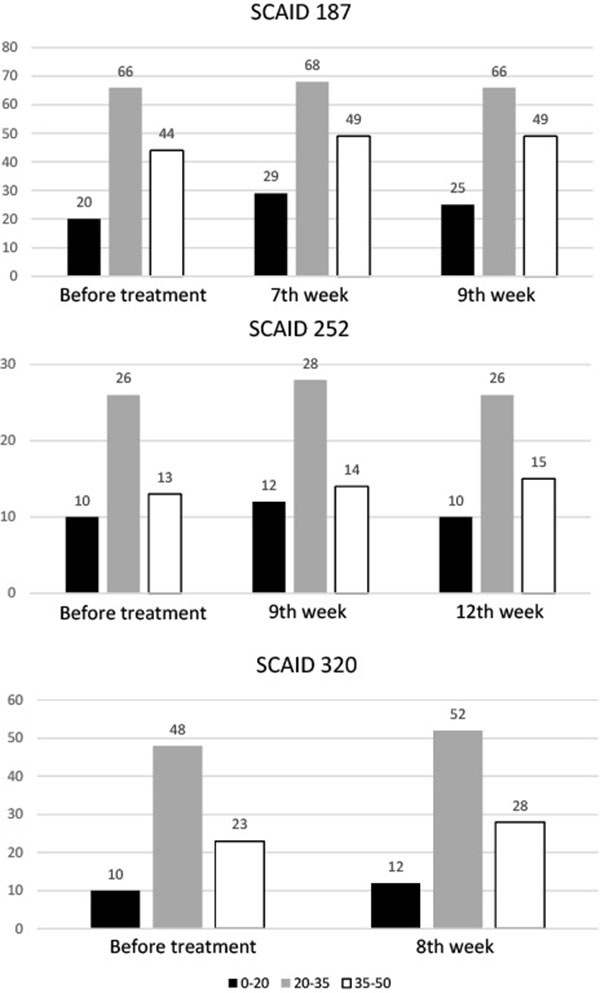 ). Total numbers of polymorphic loci (Y axis on the graphs) and frequencies of alternative alleles were counted. The polymorphic loci were grouped into 3 categories: rare mutations (0-20% of reads with alternative alleles), frequent mutations (20-35%) and common polymorphisms (35-50%). While the differences in numbers of polymorphic loci were not so great, it should be accounted that the used sequencing technique had summarized mutations in billions of sampled individual chromosomes, i.e. the graphs in Fig. (5) showed the general trends in accumulation of the most common polymorphisms leaving alone multiple individual mutations. Majority of these mutations were found in hypervariable PE-PGRS genes and in non-coding sequences except for those discussed above.
). Total numbers of polymorphic loci (Y axis on the graphs) and frequencies of alternative alleles were counted. The polymorphic loci were grouped into 3 categories: rare mutations (0-20% of reads with alternative alleles), frequent mutations (20-35%) and common polymorphisms (35-50%). While the differences in numbers of polymorphic loci were not so great, it should be accounted that the used sequencing technique had summarized mutations in billions of sampled individual chromosomes, i.e. the graphs in Fig. (5) showed the general trends in accumulation of the most common polymorphisms leaving alone multiple individual mutations. Majority of these mutations were found in hypervariable PE-PGRS genes and in non-coding sequences except for those discussed above.
 |
Fig. (5) Numbers of polymorphic loci in Mtb isolates. Black bars: rare mutations, Grey bars: frequent mutations, White bars: common polymorphisms. |
CONCLUSION
Application of FS-1 in combination with the conventional antibiotics significantly reduced the time of antibiotic therapy of MDR-TB infections from 24 months to 6-12 months with a significant reduction of the rate of post-treatment relapses of the disease. Mtb cultures isolated from the sputum samples during the treatment course showed a reversed drug resistance phenotype at least in relation to several antibiotics (Table 2). Reversion of drug resistance was considered as an important factor of the therapeutic action of FS-1. The aim of this study was to identify possible mechanisms of the drug resistance reversion by sequencing of the serial Mtb isolates obtained during the treatment course and by comparing them to the complete genome sequences of the initial MDR-TB strains. The study showed that the application of FS-1 prevented the accumulation of new drug resistance mutations in the population and reduced the initial level of drug resistance of the MDR-TB infections despite the selective pressure of antibiotics. Several canonical drug resistance mutations identified in the genomes of the initial strains remained intact in all the subsequent isolates. It was concluded that the presence of canonical mutations may not be sufficient to render an effective drug resistance. Therapy with FS-1 caused an accumulation of disruptive mutations in the phthiocerol synthase subunits and rising of the general genetic heterogeneity of the populations. Weakening of the drug resistance in consequent Mtb isolates may be explained by a disruption of the necessary genetic context by random mutations and by counter-selecting of the most resistant Mtb variants caused by an aggravation of the cumulated fitness cost. The genomic context of drug resistance may include important compensatory mutations, specific alterations in gene expression regulation and even specific DNA methylation patterns [32Heusipp, G.; Fälker, S.; Schmidt, M.A. DNA adenine methylation and bacterial pathogenesis. Int. J. Med. Microbiol., 2007, 297(1), 1-7.
[http://dx.doi.org/10.1016/j.ijmm.2006.10.002] [PMID: 17126598] ]. Additional studies should be performed to elucidate these possible mechanisms. The hypothesis of fitness cost aggravation is in line with previous scientific reports on this topic. It was demonstrated that many drug resistance mutations in prodrug activators made the mutants more sensitive to the oxidative stress caused by the iodine containing FS-1 [33Pym, A.S.; Saint-Joanis, B.; Cole, S.T. Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect. Immun., 2002, 70(9), 4955-4960.
[http://dx.doi.org/10.1128/IAI.70.9.4955-4960.2002] [PMID: 12183541] , 34Kalykova, A.; Kustova, T.; Sakipova, Z. Acute and subchronic toxicity studies of the original drug FS-1. Acta Vet. Brno, 2016, 85, 9-16.
[http://dx.doi.org/10.2754/avb201685010009] ]. To the best of our knowledge, this work was the first experimental confirmation of the drug induced antibiotic resistance reversion by the induced synergy mechanism that had been predicted theoretically in the recent publication [3Baym, M.; Stone, L.K.; Kishony, R. Multidrug evolutionary strategies to reverse antibiotic resistance. Science, 2016, 351(6268), aad3292.
[http://dx.doi.org/10.1126/science.aad3292] [PMID: 26722002] ]. Further analysis of drug resistance constraints may allow identification of targets for new antibiotics and new inducers of drug resistance reversion.
LIST OF ABBREVIATIONS
| AMI | = Amikacin Resistance. |
| CAP | = Capreomycin. |
| CSR | = Cycloserine. |
| EMB | = Ethambutol Resistance. |
| ETH | = Ethionamide Resistance. |
| FLQ | = Fluoroquinolones Resistance. |
| GMTV database | = Genome-based Mycobacterium Tuberculosis Variation Database. |
| INH | = Isoniazid Resistance. |
| KAN | = Kanamycin. |
| MDR | = Multidrug Resistance. |
| MIC | = Minimal Inhibitory Concentration. |
| Mtb | = Mycobacterium Tuberculosis. |
| OFL | = Oflaxacin. |
| PAS | = Para-Aminosalisylic Acid Resistance. |
| PZA | = Pyrazinamide Resistance. |
| RIF | = Rifampicin Resistance. |
| SCAID | = Scientific Center for Anti-Infectious Drugs. |
| SM | = Streptomycin Resistance. |
| SNP | = Single Nucleotide Polymorphism. |
| TB | = Tuberculosis. |
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Experimental procedures and sequencing were funded by the grant 0115РК00389 of the program “Study on the reversion of antibiotic resistance in pathogenic microorganisms” provided by the Industrial development and industrial safety committee of the ministry for investments and development in Kazakhstan. Bioinformatics analysis and programming for this project were funded by the grant #93664 of the National Research Foundation (NRF) of South Africa.
REFERENCES
| [1] | Tenover, F.C. Development and spread of bacterial resistance to antimicrobial agents: an overview. Clin. Infect. Dis., 2001, 33(Suppl. 3), S108-S115. [http://dx.doi.org/10.1086/321834] [PMID: 11524705] |
| [2] | Morens, D.M.; Folkers, G.K.; Fauci, A.S. The challenge of emerging and re-emerging infectious diseases. Nature, 2004, 430(6996), 242-249. [http://dx.doi.org/10.1038/nature02759] [PMID: 15241422] |
| [3] | Baym, M.; Stone, L.K.; Kishony, R. Multidrug evolutionary strategies to reverse antibiotic resistance. Science, 2016, 351(6268), aad3292. [http://dx.doi.org/10.1126/science.aad3292] [PMID: 26722002] |
| [4] | O’Neill, J. Tackling drug resistance globally: final report and recommendations. The Review on Antimicrobial Resistance, 2016. Available at: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf |
| [5] | Nouvel, L.X.; Kassa-Kelembho, E.; Dos Vultos, T.; Zandanga, G.; Rauzier, J.; Lafoz, C.; Martin, C.; Blazquez, J.; Talarmin, A.; Gicquel, B. Multidrug-resistant Mycobacterium tuberculosis, Bangui, Central African Republic. Emerg. Infect. Dis., 2006, 12(9), 1454-1456. [http://dx.doi.org/10.3201/eid1209.060361] [PMID: 17073103] |
| [6] | Cohn, D.L.; Bustreo, F.; Raviglione, M.C. Drug-resistant tuberculosis: review of the worldwide situation and the WHO/IUATLD Global Surveillance Project. Clin Infect Dis., 1997, 24(Suppl. 1) |
| [7] | Zager, E.M.; McNerney, R. Multidrug-resistant tuberculosis. BMC Infect. Dis., 2008, 8, 10. [http://dx.doi.org/10.1186/1471-2334-8-10] [PMID: 18221534] |
| [8] | Sandgren, A.; Strong, M.; Muthukrishnan, P.; Weiner, B.K.; Church, G.M.; Murray, M.B. Tuberculosis drug resistance mutation database. PLoS Med., 2009, 6(2), e2. [http://dx.doi.org/10.1371/journal.pmed.1000002] [PMID: 19209951] |
| [9] | Van, den Boogaard, J.; Kibiki, G.S.; Kisanga, E.R. New drugs against tuberculosis: problems, progress, and evaluation of agents in clinical development. Antimicrob Agents Chemother, 2009, 53(3), 849-862. |
| [10] | Mahajan, R. Bedaquiline: First FDA-approved tuberculosis drug in 40 years. Int. J. Appl. Basic Med. Res., 2013, 3(1), 1-2. [http://dx.doi.org/10.4103/2229-516X.112228] [PMID: 23776831] |
| [11] | Gupta, R.; Geiter, L.J.; Wells, C.D.; Gao, M.; Cirule, A.; Xiao, H. Delamanid for extensively drug-resistant tuberculosis. N. Engl. J. Med., 2015, 373(3), 291-292. [http://dx.doi.org/10.1056/NEJMc1415332] [PMID: 26176402] |
| [12] | Ilin, A.I.; Kulmanov, M.E. Antibacterial agent for treating infectious diseases of bacterial origin. Patent WO 2012091534 A1, 2012. |
| [13] | Jagannath, C.; Reddy, V.M.; Gangadharam, P.R. Enhancement of drug susceptibility of multi-drug resistant strains of Mycobacterium tuberculosis by ethambutol and dimethyl sulphoxide. J. Antimicrob. Chemother., 1995, 35(3), 381-390. [http://dx.doi.org/10.1093/jac/35.3.381] [PMID: 7782254] |
| [14] | Chernyaeva, E.N.; Shulgina, M.V.; Rotkevich, M.S.; Dobrynin, P.V.; Simonov, S.A.; Shitikov, E.A.; Ischenko, D.S.; Karpova, I.Y.; Kostryukova, E.S.; Ilina, E.N.; Govorun, V.M.; Zhuravlev, V.Y.; Manicheva, O.A.; Yablonsky, P.K.; Isaeva, Y.D.; Nosova, E.Y.; Mokrousov, I.V.; Vyazovaya, A.A.; Narvskaya, O.V.; Lapidus, A.L.; OBrien, S.J. Genome-wide Mycobacterium tuberculosis variation (GMTV) database: a new tool for integrating sequence variations and epidemiology. BMC Genomics, 2014, 15, 308. [http://dx.doi.org/10.1186/1471-2164-15-308] [PMID: 24767249] |
| [15] | Cohen, T.; Murray, M. Modeling epidemics of multidrug-resistant M. tuberculosis of heterogeneous fitness. Nat. Med., 2004, 10(10), 1117-1121. [http://dx.doi.org/10.1038/nm1110] [PMID: 15378056] |
| [16] | Cohen, T.; Sommers, B.; Murray, M. The effect of drug resistance on the fitness of Mycobacterium tuberculosis. Lancet Infect. Dis., 2003, 3(1), 13-21. [http://dx.doi.org/10.1016/S1473-3099(03)00483-3] [PMID: 12505028] |
| [17] | Pym, A.S.; Saint-Joanis, B.; Cole, S.T. Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect. Immun., 2002, 70(9), 4955-4960. [http://dx.doi.org/10.1128/IAI.70.9.4955-4960.2002] [PMID: 12183541] |
| [18] | Gagneux, S.; Long, C.D.; Small, P.M.; Van, T.; Schoolnik, G.K.; Bohannan, B.J. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science, 2006, 312(5782), 1944-1946. [http://dx.doi.org/10.1126/science.1124410] [PMID: 16809538] |
| [19] | Luciani, F.; Sisson, S.A.; Jiang, H.; Francis, A.R.; Tanaka, M.M. The epidemiological fitness cost of drug resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA, 2009, 106(34), 14711-14715. [http://dx.doi.org/10.1073/pnas.0902437106] [PMID: 19706556] |
| [20] | Bloch, H.; Segal, W. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J. Bacteriol., 1956, 72(2), 132-141. [PMID: 13366889] |
| [21] | World Health Organization. Interim Policy Guidance on Drug Susceptibility Testing (DST) of Second-Line Anti-Tuberculosis Drugs World Health Organization Document, 2008. WHO/HTM/TB/2008.392 |
| [22] | Krüüner, A.; Yates, M.D.; Drobniewski, F.A. Evaluation of MGIT 960-based antimicrobial testing and determination of critical concentrations of first- and second-line antimicrobial drugs with drug-resistant clinical strains of Mycobacterium tuberculosis. J. Clin. Microbiol., 2006, 44(3), 811-818. [http://dx.doi.org/10.1128/JCM.44.3.811-818.2006] [PMID: 16517859] |
| [23] | Murray, M.G.; Thompson, W.F. Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res., 1980, 8(19), 4321-4325. [http://dx.doi.org/10.1093/nar/8.19.4321] [PMID: 7433111] |
| [24] | Ilin, A.I.; Kulmanov, M.E.; Korotetskiy, I.S.; Akhmetova, G.K.; Lankina, M.V.; Shvidko, S.V.; Reva, O.N. Complete genome sequence of multi-drug resistant clinical isolate Mycobacterium tuberculosis 187.0 used to study an effect of drug susceptibility reversion by a new medicinal drug FS-1. Genome Announc., 2015, 3(6), e01272-e15. [http://dx.doi.org/10.1128/genomeA.01272-15] [PMID: 26543112] |
| [25] | Yang, J.; Shikano, T.; Li, M-H.; Merilä, J. Genome-wide linkage disequilibrium in nine-spined stickleback populations. G3 (Bethesda), 2014, 4(10), 1919-1929. [http://dx.doi.org/10.1534/g3.114.013334] [PMID: 25122668] |
| [26] | Darling, A.C. Mau. B.; Blattner, F.D.; Perna, N.T. Mauve: multiple alignment of conserved genomic sequences with rearrangements. Genet. Res., 2004, 14(7), 1394-1403. [http://dx.doi.org/10.1101/gr.2289704] |
| [27] | Merker, M.; Blin, C.; Mona, S.; Duforet-Frebourg, N.; Lecher, S.; Willery, E.; Blum, M.G.; Rüsch-Gerdes, S.; Mokrousov, I.; Aleksic, E.; Allix-Béguec, C.; Antierens, A.; Augustynowicz-Kopeć, E.; Ballif, M.; Barletta, F.; Beck, H.P.; Barry, C.E., III; Bonnet, M.; Borroni, E.; Campos-Herrero, I.; Cirillo, D.; Cox, H.; Crowe, S.; Crudu, V.; Diel, R.; Drobniewski, F.; Fauville-Dufaux, M.; Gagneux, S.; Ghebremichael, S.; Hanekom, M.; Hoffner, S.; Jiao, W.W.; Kalon, S.; Kohl, T.A.; Kontsevaya, I.; Lillebæk, T.; Maeda, S.; Nikolayevskyy, V.; Rasmussen, M.; Rastogi, N.; Samper, S.; Sanchez-Padilla, E.; Savic, B.; Shamputa, I.C.; Shen, A.; Sng, L.H.; Stakenas, P.; Toit, K.; Varaine, F.; Vukovic, D.; Wahl, C.; Warren, R.; Supply, P.; Niemann, S.; Wirth, T. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat. Genet., 2015, 47(3), 242-249. [http://dx.doi.org/10.1038/ng.3195] [PMID: 25599400] |
| [28] | Fraaije, M.W.; Kamerbeek, N.M.; Heidekamp, A.J.; Fortin, R.; Janssen, D.B. The prodrug activator EtaA from Mycobacterium tuberculosis is a Baeyer-Villiger monooxygenase. J. Biol. Chem., 2004, 279(5), 3354-3360. [http://dx.doi.org/10.1074/jbc.M307770200] [PMID: 14610090] |
| [29] | Yu, J.; Tran, V.; Li, M.; Huang, X.; Niu, C.; Wang, D.; Zhu, J.; Wang, J.; Gao, Q.; Liu, J. Both phthiocerol dimycocerosates and phenolic glycolipids are required for virulence of Mycobacterium marinum. Infect. Immun., 2012, 80(4), 1381-1389. [http://dx.doi.org/10.1128/IAI.06370-11] [PMID: 22290144] |
| [30] | Torrey, H.L.; Keren, I.; Via, L.E.; Lee, J.S.; Lewis, K. High persister mutants in Mycobacterium tuberculosis. PLoS One, 2016, 11(5) e0155127. [http://dx.doi.org/10.1371/journal.pone.0155127] [PMID: 27176494] |
| [31] | Williamson, M.P.; Keren, I.; Via, L.E. The structure and function of proline-rich regions in proteins. Biochem. J., 1994, 297(Pt 2), 249-260. [http://dx.doi.org/10.1042/bj2970249] [PMID: 8297327] |
| [32] | Heusipp, G.; Fälker, S.; Schmidt, M.A. DNA adenine methylation and bacterial pathogenesis. Int. J. Med. Microbiol., 2007, 297(1), 1-7. [http://dx.doi.org/10.1016/j.ijmm.2006.10.002] [PMID: 17126598] |
| [33] | Pym, A.S.; Saint-Joanis, B.; Cole, S.T. Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect. Immun., 2002, 70(9), 4955-4960. [http://dx.doi.org/10.1128/IAI.70.9.4955-4960.2002] [PMID: 12183541] |
| [34] | Kalykova, A.; Kustova, T.; Sakipova, Z. Acute and subchronic toxicity studies of the original drug FS-1. Acta Vet. Brno, 2016, 85, 9-16. [http://dx.doi.org/10.2754/avb201685010009] |