- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




The Open Virology Journal
(Discontinued)
ISSN: 1874-3579 ― Volume 15, 2021
The microRNA Transcriptome of Human Cytomegalovirus (HCMV)
Mesfin K Meshesha 1, §, Isana Veksler-Lublinsky 2, §, Ofer Isakov 3, Irit Reichenstein 1, Noam Shomron 3, Klara Kedem 2, Michal Ziv-Ukelson 2, Zvi Bentwich 1, Yonat Shemer Avni *, 1
Abstract
The purpose of the present study was to characterize the microRNA transcriptome (miRNAome) of the human cytomegalovirus (HCMV or HHV5). We used deep sequencing and real time PCR (qPCR) together with bioinformatics to analyze the pattern of small RNA expression in cells infected with low-passage isolates of HCMV as well as in plasma and amniotic fluid. We report here on the discovery of four new precursors and ten new miRNAs as well as eleven microRNA-offset-RNAs (moRs) that are all encoded by HCMV. About eighty percent of the total HCMV reads were perfectly mapped to HCMV miRNAs, strongly suggestive of their important biological role that in large remains still to be defined and characterized. Taken altogether, the results of this study demonstrate the power and usefulness of the combined bioinformatics/biological approach in discovering additional important members of HCMV- encoded small RNAs and can be applied to the study of other viruses as well.
Article Information
Identifiers and Pagination:
Year: 2012Volume: 6
First Page: 38
Last Page: 48
Publisher Id: TOVJ-6-38
DOI: 10.2174/1874357901206010038
Article History:
Received Date: 25/1/2012Revision Received Date: 22/2/2012
Acceptance Date: 23/2/2012
Electronic publication date: 11/4/2012
Collection year: 2012
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Department of Virology and Molecular Genetics, Faculty of Health Sciences, Ben Gurion University of the Negev, Beer Sheva 84105, Israel; Tel: +972 8 6403133; E-mail: yonat@bgu.ac.il§ Equal contribution.
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 25-1-2012 |
Original Manuscript | The microRNA Transcriptome of Human Cytomegalovirus (HCMV) | |
INTRODUCTION
Human cytomegalovirus (HCMV or HHV5) is ubiquitous and a highly prevalent human pathogen, usually acquired at an early age. HCMV belongs to the beta subfamily of herpesviridae characterized by linear dsDNA. Following primary infection, the virus establishes life-long latent infection with episodes of reactivation, mainly in the immune compromised host. The tissue distribution of HCMV is largely dependent on the host immune status, as well [1 Fields BN, Knipe DM, Howley PM, Griffin DE, Eds. Fields virology. 4th. Philadelphia: Lippincott Williams & Wilkins 2001.]. A substantial part of its genetic information is dedicated to establish the lytic and latent stages of the virus life cycle and to control the immune responses of the host. Three sequential studies [2 Dunn W, Trang P, Zhong Q, Yang E, van Belle C, Liu F. Humancytomegalovirus expresses novel microRNAs during productive viralinfection Cell Microbiol 2005; 7(11): 1684-95.-4 Pfeffer S, Sewer A, Lagos-Quintana M, et al. Identification ofmicroRNAs of the herpesvirus family Nat Methods 2005; 2(4 ): 269-76.] demonstrated that the HCMV genome, as other members of its herpsviridae family, encode for microRNAs (miRNAs). These small RNAs were shown to participate in the complex regulation of host cell metabolism and to assist in establishing latency and immune evasion [5 Grey F, Meyers H, White EA, Spector DH, Nelson J. A humancytomegalovirus-encoded microRNA regulates expression of multipleviral genes involved in replication PLoS Pathog 2007; 3(11): e163.,6 Stern-Ginossar N, Elefant N, Zimmermann A, et al. Host immunesystem gene targeting by a viral miRNA Science 2007; 317(5836): 376-81.].
So far, 17 miRNAs originating from 11 precursors have been found to be encoded by HCMV (see review [7 Tuddenham L, Pfeffer S. Roles and regulation of microRNAs incytomegalovirus infection Biochim Biophys Acta 2011; 1809(11-12): 613-22.]). Unlike most of the herpesviridae, the miRNAs of HCMV are scattered along both strands of its genome, and are expressed through productive infection. Five pre-miRNAs originate from intergenic regions, another four are transcribed antisense to ORFs of annotated genes and mir-UL36 is encoded within an intron of UL36 gene (see review [8 Dhuruvasan K, Sivasubramanian G, Pellett PE. Roles of host and viralmicroRNAs in human cytomegalovirus biology Virus Res 2011; 157(2): 180-92.]).
The use of massive parallel sequencing (deep sequencing, DS), for characterizing miRNAs, has opened a broader opportunity to discover new miRNAs that are expressed at low levels and to characterize the relative expression of known miRNAs [9 Zhu JY, Pfuhl T, Motsch N, et al. Identification of novel Epstein-Barrvirus microRNA genes from nasopharyngeal carcinomas J Virol 2009; 83(7): 3333-41.]. So far, deep sequencing of herpes viruses has revealed the complex nature of miRNA generation, with variability in length and sequence giving rise to isoforms of miRNAs (for example, [10 Umbach JL, Cullen BR. In-depth analysis of Kaposi's sarcomaassociatedherpesvirus microRNA expression provides insights into themammalian microRNA-processing machinery J Virol 2010; 84(2): 695-703.,11 Chen SJ, Chen GH, Chen YH, et al. Characterization of Epstein-Barrvirus miRNAome in nasopharyngeal carcinoma by deep sequencing PLoS One 2010; 5(9): e12745.]).
The presence of diverse short RNAs - miRNA offset RNAs (moRs) was discovered in Ciona intestinalis [12 Shi W, Hendrix D, Levine M, Haley B. A distinct class of small RNAsarises from pre-miRNA-proximal regions in a simple chordate NatStruct Mol Biol 2009; 16(2): 183-9.], in human cells [13 Langenberger D, Bermudez-Santana C, Hertel J, Hoffmann S, Khaitovich P, Stadler PF. Evidence for human microRNA-offset RNAsin small RNA sequencing data Bioinformatics 2009; 25(18): 2298-301.], in KSHV (or HHV8) [10 Umbach JL, Cullen BR. In-depth analysis of Kaposi's sarcomaassociatedherpesvirus microRNA expression provides insights into themammalian microRNA-processing machinery J Virol 2010; 84(2): 695-703.] and in HSV1 and 2 [14 Jurak I, Kramer MF, Mellor JC, et al. Numerous conserved anddivergent microRNAs expressed by herpes simplex viruses 1 and 2 J Virol 2010; 84(9): 4659-72.]. moRs are typically found in the vicinity of known or predicted miRNAs, in the 5' or 3'-end of the pre-miRNA [12 Shi W, Hendrix D, Levine M, Haley B. A distinct class of small RNAsarises from pre-miRNA-proximal regions in a simple chordate NatStruct Mol Biol 2009; 16(2): 183-9.,13 Langenberger D, Bermudez-Santana C, Hertel J, Hoffmann S, Khaitovich P, Stadler PF. Evidence for human microRNA-offset RNAsin small RNA sequencing data Bioinformatics 2009; 25(18): 2298-301.,15 Bortoluzzi S, Biasiolo M, Bisognin A. MicroRNA-offset RNAs(moRNAs): by-product spectators or functional players? Trends Mol Med 2011; 17(9): 473-.]. However, the majority of moRs are derived from the 5’-side of the stem. This is independent of whether the 3’- or 5’- side is predominantly processed into miRNAs. Furthermore, it was reported that in some cases the abundance of moRs exceeds the abundance of the mature miRNA derived from the same pre-miRNA (in Ciona intestinalis [12 Shi W, Hendrix D, Levine M, Haley B. A distinct class of small RNAsarises from pre-miRNA-proximal regions in a simple chordate NatStruct Mol Biol 2009; 16(2): 183-9.] and in human monocytic leukemia cell line (THP-1) [16 Taft RJ, Simons C, Nahkuri S, et al. Nuclear-localized tiny RNAs areassociated with transcription initiation and splice sites in metazoans Nat Struct Mol Biol 2010; 17(8): 1030-4.]), suggesting that moRs and miRNA biogenesis may be linked but are not interdependent [16 Taft RJ, Simons C, Nahkuri S, et al. Nuclear-localized tiny RNAs areassociated with transcription initiation and splice sites in metazoans Nat Struct Mol Biol 2010; 17(8): 1030-4.]. Though the biological function of this species of small RNAs is not yet clear, the current hypothesis is that moRs are a new class of regulatory short RNA. Furthermore, Taft et al. [16 Taft RJ, Simons C, Nahkuri S, et al. Nuclear-localized tiny RNAs areassociated with transcription initiation and splice sites in metazoans Nat Struct Mol Biol 2010; 17(8): 1030-4.] showed that moRs are enriched in the nucleus of human monocytic leukemia cells (THP-1), which could be associated with transcription.
Even though the size of its genome is larger than the genome of other members of this family; e.g., EBV (or HHV4) and KSHV, the number of miRNAs described so far for HCMV is smaller (http://www.mirbase.org/). In the current study we have applied a combined approach using DS and qPCR together with bioinformatics tools to study the miRNA transcriptome (miRNAome) of HCMV. The results of this study have demonstrated new HCMV encoded precursors, new miRNAs as well as new moRs, prompting this report. Furthermore, we were able to detect all the newly discovered HCMV-miRNA not only in HCMV infected fibroblasts but also in plasma and amniotic fluid during HCMV infection.
MATERIALS AND METHODS
Cells and viruses
Human foreskin fibroblast (HFF) cells (kindly provided by Dr. Kra-Oz, Haifa, Israel) were cultured in BIO-AMF complete media (Biological Industries, BeitHaemek, Israel) and used to propagate two clinical isolates and the laboratory strain AD169 of HCMV.
HFF cells were infected with two clinical isolates and AD169 laboratory strain, at multiplicity of infection (MOI) of two.
RNA extraction
Total RNA was extracted 96 hours post infection using EZ-RNA II isolation kit (Biological Industries, Beit Haemek, Israel), according to the manufacturer's protocol. Aliquots of total RNA were used directly for miRNA quantification using qPCR or subjected to small RNA library construction.
Deep Sequencing
DS was carried out at the Tel-Aviv University Genome High-Throughput Sequencing Laboratory following Illumina’s Small RNA sample preparation protocol v1.5 (TruSeq Small RNA Sample Prep Kits, Illumina) for generating small RNA libraries directly from total RNA and for sequencing. miRNA libraries were sequenced on the Illumina Cluster Station and Genome Analyzer II following the manufacturer’s protocol.
qPCR analysis of HCMV miRNAs
cDNA libraries were prepared using the miScript Reverse Transcription Kit (QIAGEN, Hilden, Germany). Briefly, 1 µg of total RNA (from hCMV infected and uninfected cells) were poly-adenylated by poly (A) polymerase and converted into cDNA by reverse transcriptase with oligo-dT in a single step, at 37oC for 1 hr. and 95oC for 5 min using the miScript Reverse Transcription kit (QIAGEN, Hilden, Germany). The cDNA libraries were used in the qPCR analysis for miRNAs using the miScript SYBR Green PCR Kit (QIAGEN, Hilden, Germany) with a forward specific miRNA primer (sequences were taken from the DS analysis, Metabion Martinsried, Germany) and the miScript Universal reverse primer. Forward primers with high G/C content at the 3' end, were extended with one or two 'A' nt at their 3'-end, see Table 1 for list of all primers used in this study. qPCR reactions were performed using the Light Cycler 480 system (Roche Applied Science, Manheim, Germany) and the amplicons were run on a 2% (w/v) agarose gel. Only CMV-miRNAs that were qPCR negative in the uninfected library, and with the correct size of the amplicon were considered as validated.
Analysis of the deep-sequencing data
Sequencing results were first filtered by removing the adaptor sequence and keeping reads with a size of more than 18 nt. The filtered reads were then aligned to the reference Human herpesvirus 5 (HCMV) genome (NC_006273.2) using the BWA program (0.5.8a version downloaded from http://sourceforge.net/projects/bio-bwa/files/), forbidding mismat-ches in the alignment. A genomic map of the virus sequence was built with annotation of its known pre-miRNAs and mature miRNAs (downloaded from miRBase, v.17.0 http://www.mirbase.org/). The results of the BWA were used to assign, to each position in the genome, the reads which start at that position. Based on this, each position was annotated with a read count indicating the number of reads starting at this position (http://www.cs.bgu.ac.il/~vaksler/PROJ/GenomeMapper.htm ).
Identification of new HCMV miRNAs within known precursors
We inspected the known pre-miRNAs in the genomic map in which only one arm of the precursor contains a known miRNA and checked the number of reads mapped to the other arm. Whenever the read count was above 25, the corresponding sequence was considered as a candidate miRNA and was further checked by qPCR.
Identification of new HCMV precursors and miRNAs
We traversed each position in the genomic map in order to identify peaks that are putative start indices of miRNAs. These positions comply with the following criteria: (1) a read count of at least 25, (2) at least 70% of these reads have length ranging between 20-23 nt, and (3) the neighboring positions in the range 6-15 nt on both sides of the considered position all have a read count smaller than 10 (this to guarantee that there are no abundant small RNAs starting in the middle, or too closely above, a putative miRNA, and to take into account the allowed offsets of minus 5 to 5, see Results). In order to further take into account the secondary structure of the potential miRNAs starting at each putative start position, two windows of 100nt in size were considered such that the putative miRNA sequence could participate in either arm of the putative stem. The predicted structures were obtained via MFOLD [17 Zuker M. Mfold web server for nucleic acid folding and hybridizationprediction Nucleic Acids Res 2003; 31(13): 3406-15.]. The windows whose folding yielded stem-loop hairpins and met the following criteria were considered typical precursors: the sequence with high read count falls in the stem of the hairpin and the free-folding energy is lower than -25 kcal/mol. In addition, the pairing number of the mature sequence within the hairpin should be higher than 14 nts, and the maximal bulge should be less than 5 nts. The putative miRNAs from predicted precursors were further checked by real time PCR (qPCR).
Identification of HCMV encoded miRNA offset RNAs (moRs)
miRNA offset RNAs (moRs) are gaining popularity in the field of small RNAs. We investigated the presence of these 19-20 nt RNAs in our DS data. To find potential moRs we scanned all known and predicted precursors and checked if there were peaks of read counts with starting position approximately 18-20 nt before the 5p-miRNA or at the end of the 3p-miRNA. In addition, we required that the sought moRs be part of a stem in the putative folding.
RESULTS
Deep sequencing
The aim of our study was to characterize the miRNAome of HCMV. In pursue of known and new miRNAs encoded by HCMV we applied high throughput DS analysis to small RNA prepared from HCMV infected HFF cells.
Among the 16,837,317 reads obtained from the deep-sequencing, 1,015,588 (6%) were perfectly matched to the HCMV genome. These latter reads were assigned to the genomic map of the HCMV using the results from BWA (http://www.cs.bgu.ac.il/~vaksler/PROJ/GenomeMapper.htm). From now on we refer just to these latter reads. 812,477 (80%) of the reads were mapped to the known HCMV miRNAs, 35,727 (4%) of the reads were mapped to new miRNAs, and 7903 (0.7%) of the reads were mapped to moRs. The read count of a specific miRNA includes all the sequences such that their start position deviates from that of the miRBase sequence (our 0-offset) by an offset of -5 to 5 nt. For new miRNAs and moRs, the deviation is from the most abundant sequence (which is our 0-offset).
Fig. (1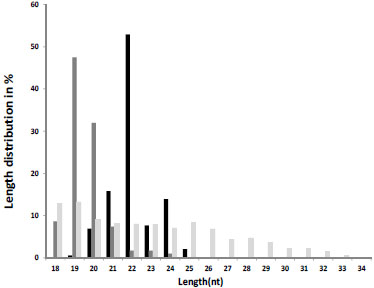 ) illustrates the length distribution of reads that were mapped to miRNAs (black), moRs (dark grey), and other small RNAs (light grey). As known from the literature [18 Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNAannotation RNA 2003; 9(3): 277-9.] the length of miRNAs is ~21-22nt. Our DS supports this fact since more than half of the reads mapped to the HCMV miRNAs (known and new) had a length of 22 nt, and ranged from 19 nt to 25 nt. moRs peaked at length of 19-20 nt. The length of the rest of the reads was distributed between 18 to 32nt.
) illustrates the length distribution of reads that were mapped to miRNAs (black), moRs (dark grey), and other small RNAs (light grey). As known from the literature [18 Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNAannotation RNA 2003; 9(3): 277-9.] the length of miRNAs is ~21-22nt. Our DS supports this fact since more than half of the reads mapped to the HCMV miRNAs (known and new) had a length of 22 nt, and ranged from 19 nt to 25 nt. moRs peaked at length of 19-20 nt. The length of the rest of the reads was distributed between 18 to 32nt.
The dominant sequence variants were compared to the consensus data in the miRBase website, and the distribution of the offsets of miRNA start points (on the 5' end) and end points (on the 3' end) from the 0-offset was analyzed. The variation in start points at the 5'-end (Fig. 2A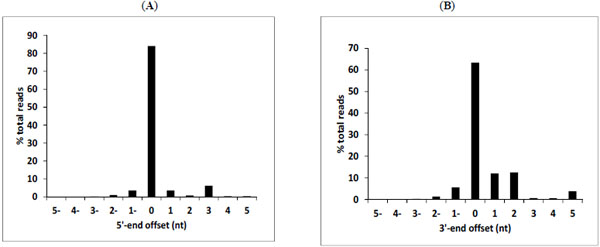 ) was smaller than the variation of end points at the 3'-end (Fig. 2B). The frequency of 5' and 3' ends variation was 15% and 65% respectively. The extent of length and offset variations were observed previously in EBV DS analysis as well, and also in human and mouse miRNAs [11 Chen SJ, Chen GH, Chen YH, et al. Characterization of Epstein-Barrvirus miRNAome in nasopharyngeal carcinoma by deep sequencing PLoS One 2010; 5(9): e12745.,19 Morin RD, O'Connor MD, Griffith M, et al. Application of massivelyparallel sequencing to microRNA profiling and discovery in humanembryonic stem cells Genome Res 2008; 18(4): 610-21.-21 Reid JG, Nagaraja AK, Lynn FC, et al. Mouse let-7 miRNApopulations exhibit RNA editing that is constrained in the 5'-seed/cleavage/anchor regions and stabilize predicted mmu-let-7a:mRNAduplexes Genome Res 2008; 18(10): 1571-81.].
) was smaller than the variation of end points at the 3'-end (Fig. 2B). The frequency of 5' and 3' ends variation was 15% and 65% respectively. The extent of length and offset variations were observed previously in EBV DS analysis as well, and also in human and mouse miRNAs [11 Chen SJ, Chen GH, Chen YH, et al. Characterization of Epstein-Barrvirus miRNAome in nasopharyngeal carcinoma by deep sequencing PLoS One 2010; 5(9): e12745.,19 Morin RD, O'Connor MD, Griffith M, et al. Application of massivelyparallel sequencing to microRNA profiling and discovery in humanembryonic stem cells Genome Res 2008; 18(4): 610-21.-21 Reid JG, Nagaraja AK, Lynn FC, et al. Mouse let-7 miRNApopulations exhibit RNA editing that is constrained in the 5'-seed/cleavage/anchor regions and stabilize predicted mmu-let-7a:mRNAduplexes Genome Res 2008; 18(10): 1571-81.].
Known HCMV miRNAs
HCMV encodes 17 known mature miRNAs from 11 precursors [22 Kozomara A, Griffiths-Jones S. miRBase: integrating microRNAannotation and deep-sequencing data Nucleic Acids Res 2011; 39(Database issue
): D152-7.]. Sequence information and the read counts for these miRNAs are summarized in Table 2. Interestingly, we found that most of the HCMV miRNAs had many reads, some reaching 100s of thousands (Table 2 and Fig. 3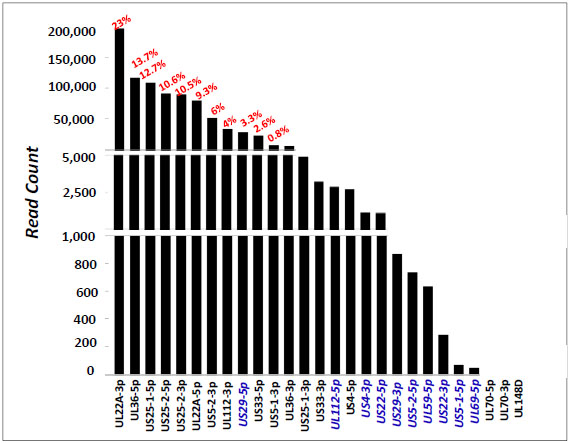 ). Thirteen of these miRNAs were found in the sample with a read count of above 3000 reads. The relative abundance of the known miRNAs is depicted in Fig. (3): miR-UL22A-3p had the highest read count (196,688), followed by miR-UL36-5p (116,669) and miR-US25-1-5p (108,624), constituting 23%, 13.7% and 12.7%, respectively, out of all reads mapped to HCMV miRNAs.
). Thirteen of these miRNAs were found in the sample with a read count of above 3000 reads. The relative abundance of the known miRNAs is depicted in Fig. (3): miR-UL22A-3p had the highest read count (196,688), followed by miR-UL36-5p (116,669) and miR-US25-1-5p (108,624), constituting 23%, 13.7% and 12.7%, respectively, out of all reads mapped to HCMV miRNAs.
Reads for mature miRNAs of mir-UL70-1 and mir-UL148D-1 were not found in our DS. Interestingly, all the reads that were mapped to miR-US4-5p were in +5 offset from the miRBase published sequence.
With respect to already described miRNA biogenesis, we observed that the miRNA/miRNA* designation in the miRBase annotation is consistent with the analysis of our DS data with a single exception of mir-UL22A-1. As opposed to the miRBase and the qPCR (data not shown), we found that mir-UL22A-3p, which has been designated as the star (*) form of this miRNA in the miRBase annotation, had a much higher sequence-read abundance than miR-UL22A-5p (see Table 2).
This alternation in expression of mature miRNAs within their precursor as well as the finding that some miRNAs were not detected by the DS technique while they were detected by another approach, e.g., the qPCR, might be attributed to technical difficulties of the DS such as ligation and preferences in PCR amplification.
The new HCMV miRNAs within known precursors
Four previously unreported miRNAs, that originated from known pre-miRNAs [3 Grey F, Antoniewicz A, Allen E, et al. Identification andcharacterization of human cytomegalovirus-encoded microRNAs J Virol 2005; 79(18): 12095-9.,4 Pfeffer S, Sewer A, Lagos-Quintana M, et al. Identification ofmicroRNAs of the herpesvirus family Nat Methods 2005; 2(4 ): 269-76.], were revealed by the DS: miR-UL112-5p, miR-US4-3p, miR-US5-1-5p, and miR-US5-2-5p, (2865, 1145, 60, and 733 reads, respectively, see Table 3A and Fig. 3). These miRNAs had a relatively low read count. Three of these miRNAs were confirmed by qPCR (Fig. 4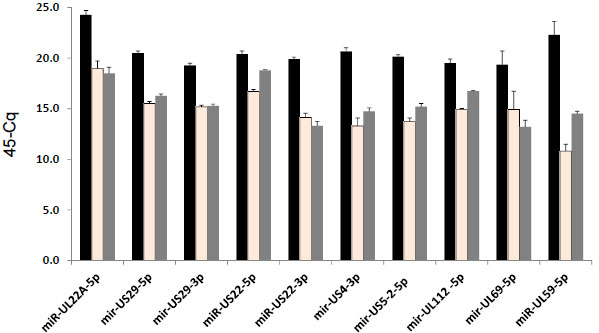 ), and thus were considered as new HCMV miRNAs. In contrast, miR-US5-1-5p had a different TM (in the TM analysis of the qPCR) and a larger size of amplicons than the expected size of a miRNA.
), and thus were considered as new HCMV miRNAs. In contrast, miR-US5-1-5p had a different TM (in the TM analysis of the qPCR) and a larger size of amplicons than the expected size of a miRNA.
New HCMV precursors and miRNAs
Using the method described above, we identified 13 precursors on both strands of the genome, nine of them are known precursors and four are new. The mature miRNAs originating from the latter precursors (see Fig. 5 ), were further checked by qPCR. Six mature miRNAs from four precursors, mir-US29-1, mir-US22-1, mir-UL69-1 and mir-UL59-1, were confirmed by qPCR (Table 3B and Fig. 4). Furthermore, we also detected these six new miRNAs in RNA extracted from plasma and amniotic fluid samples taken from HCMV infected patients using real-time PCR (data not shown). Fig. (6
), were further checked by qPCR. Six mature miRNAs from four precursors, mir-US29-1, mir-US22-1, mir-UL69-1 and mir-UL59-1, were confirmed by qPCR (Table 3B and Fig. 4). Furthermore, we also detected these six new miRNAs in RNA extracted from plasma and amniotic fluid samples taken from HCMV infected patients using real-time PCR (data not shown). Fig. (6 ) depicts the genomic locations of all these new miRNAs, along with the known miRNAs.
) depicts the genomic locations of all these new miRNAs, along with the known miRNAs.
The correct size of the amplicons of the mature miRNA was analyzed by TM-calling (Light Cycler 480) and on agarose gel.
mir-UL59-1 and mir-UL69-1 are encoded within an intergenic region between genes UL57 and UL69. Even though the 3p arm of mir-UL59 was detectable by the DS, we could not confirm it by qPCR. Both the 5p arms of these two miRNA precursors are readily detectable in HFF cells infected by clinical isolates and strain AD169 (Fig. 4) as well as in plasma and amniotic fluid specimens obtained from HCMV infected patients (data not shown).
The mir-US22-1 precursor, which contains two mature miRNAs, is transcribed from the complementary strand to gene US22, and thus the 5p and 3p mature miRNAs are likely candidates to down regulate US22 expression. (This phenomenon is similar to that observed in other previously known miRNAs (UL70-1, UL112-1, UL148D, US33-1) [3 Grey F, Antoniewicz A, Allen E, et al. Identification andcharacterization of human cytomegalovirus-encoded microRNAs J Virol 2005; 79(18): 12095-9., 4 Pfeffer S, Sewer A, Lagos-Quintana M, et al. Identification ofmicroRNAs of the herpesvirus family Nat Methods 2005; 2(4 ): 269-76.].
mir-US29 that has been predicted before [3 Grey F, Antoniewicz A, Allen E, et al. Identification andcharacterization of human cytomegalovirus-encoded microRNAs J Virol 2005; 79(18): 12095-9.], is located within the US29 gene and is transcribed antisense to the previously known mir-US33-1 (Fig. 6). The phenomenon that two precursors are transcribed from both strands of a genomic region has been previously reported in Drosophila [23 Tyler DM, Okamura K, Chung WJ, et al. Functionally distinctregulatory RNAs generated by bidirectional transcription andprocessing of microRNA loci Genes Dev 2008 ; 22(1): 26-36.], HSV-1 [14 Jurak I, Kramer MF, Mellor JC, et al. Numerous conserved anddivergent microRNAs expressed by herpes simplex viruses 1 and 2 J Virol 2010; 84(9): 4659-72.] and in KSHV [10 Umbach JL, Cullen BR. In-depth analysis of Kaposi's sarcomaassociatedherpesvirus microRNA expression provides insights into themammalian microRNA-processing machinery J Virol 2010; 84(2): 695-703.].
Conservation with Chimpanzee cytomegalovirus (CCMV)
In order to further strengthen the validity of the new HCMV miRNAs that were found by DS and confirmed by qPCR, we looked for sequence homology between the pre-miRNAs of HCMV and the Chimpanzee cytomegalovirus (CCMV), the closest relative of HCMV in herpesvirus β family [24 Davison AJ, Dolan A, Akter P, et al. The human cytomegalovirusgenome revisited: comparison with the chimpanzee cytomegalovirus genome J Gen Virol 2003; 84(Pt 1): 17-28.]. We could find similarity of 100% and 69%, respectively, between pre-miRNAs mir-US29-1 and mir-US22-1 of HCMV and the CCMV sequences. Both corresponding sequences in CCMV could be folded into hairpin secondary structures (data not shown). No homologous sequence for mir-UL59 and mir-UL69 were found in the CCMV genome that could fold into a hairpin secondary structure.
We also checked sequence homology between the mature miRNAs of HCMV and sequences of CCMV. Both miRNAs: miR-US29-5p and miR-US29-3p were fully conserved in CCMV. The seed of miR-US5-2-5p is fully conserved in CCMV, while the seeds of miR-US4-3p, miR-US22-5p, miR-US22-3p and miR-UL112-5p show a single nucleotide mismatch with CCMV.
HCMV encoded miRNA offset RNAs (moRs).
Up to this date no moRs of HCMV have yet been reported. In this work we identify 11 moRs, denoted by moR-UL22A-1-5p, moR-UL22A-1-3p, moR-UL112-5p, moR-US4-1-5p, moR-US4-1-3p, moR-US5-1-5p, moR-US5-2-5p, moR-US25-1-5p, moR-US33-1-5p, moR-US22-1-5p, and moR-US29-1-5p. In Fig. (5A, B) we present the moRs of HCMV within the secondary structure of their precursors. We also list in the figure the number of reads of the moRs. The number of reads of moRs ranged between tens to thousands. The high abundance of some of the moRs suggests that they might have important biological functions. In order to further investigate these small RNAs we selected four of the most abundant moRs: moR-US-5-2-5p, moR-UL22A-1-5p, moR-UL112-5p, and moR-US4-5p for qPCR expression analysis in our two low-passage clinical isolates and strain AD169. The qPCR detected three out of the four moRs: moR-US-5-2-5p, moR-UL112-5p, and moR-US4-5p (Fig. 7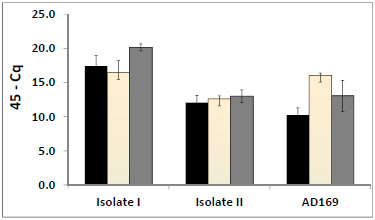 ) in all the three HCMV isolates. Size analysis of qPCR products by TM calling and gel electrophoresis confirmed their size. In spite of its high read count, the qPCR failed to detect moR-UL22A-1-5p. The fact that HCMV moRs are expressed in authentic infection, and their consistent expression in various isolates, strongly suggests that these small RNAs have important biological roles in HCMV infection.
) in all the three HCMV isolates. Size analysis of qPCR products by TM calling and gel electrophoresis confirmed their size. In spite of its high read count, the qPCR failed to detect moR-UL22A-1-5p. The fact that HCMV moRs are expressed in authentic infection, and their consistent expression in various isolates, strongly suggests that these small RNAs have important biological roles in HCMV infection.
DISCUSSION
We report here on the discovery of several new HCMV- encoded miRNA precursors, new mature miRNAs and miRNA-offset RNAs (moRs). These observations are the result of an integrated approach that combined bioinformatic analysis with high throughput sequencing technology. We believe that this presents a very powerful tool that, if applied widely for the study of the miRNAome of any organism, and particularly of other viruses, will be able to reveal new members that have not been found before. Furthermore, this approach is able to provide a better and more detailed understanding of small RNA expression and miRNA associated short RNAs.
We have used the Ambros et al. [18 Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNAannotation RNA 2003; 9(3): 277-9.] criteria for defining the putative new miRNAs that we discovered. These criteria include: A) detection of a distinct ~21-22-nt RNA transcript, originating from cDNA library of small RNA sample; B) sequences that match precisely to HCMV genome; C) prediction of potential fold-back precursor that contains the ~21-22-nt sequence within one of the arms of the hairpin, with folding free energy lower than -25 kcal; and D) phylogenetic conservation with Chimpanzee cytomegalo-virus.
Through combined bioinformatics prediction and small RNA cloning, previous studies have identified 17 miRNAs of HCMV that are processed from 11 precursors [2 Dunn W, Trang P, Zhong Q, Yang E, van Belle C, Liu F. Humancytomegalovirus expresses novel microRNAs during productive viralinfection Cell Microbiol 2005; 7(11): 1684-95.-4 Pfeffer S, Sewer A, Lagos-Quintana M, et al. Identification ofmicroRNAs of the herpesvirus family Nat Methods 2005; 2(4 ): 269-76.]. Our DS analysis enabled us to detect and confirm six new miRNAs processed from four new precursors, and four new miRNAs from already described precursors. Altogether, we have found 24 mature forms of miRNAs originating from thirteen precursors. During the time that this manuscript was prepared for submission Stark et al. article appeared in press [25 Stark TJ, Arnold JD, Spector DH, Yeo GW. High-resolution profilingand analysis of viral and host small RNAs during humancytomegalovirus infection J Virol 2011 Oct 19; ] reporting the discovery of some of our newly found miRNAs, which lends further support to our own findings
Several HCMV encoded miRNAs were found to be highly expressed, accounting for 80% of the total number of reads mapped to the HCMV genome. This high level of viral encoded miRNA expression has been reported in Marek's disease virus [26 Yao Y, Zhao Y, Xu H, et al. MicroRNA profile of Marek's diseasevirus-transformed T-cell line MSB-1: predominance of virus-encoded microRNAs J Virol 2008; 82(8): 4007-15.] but has not been observed before in other members of the herpes virus family [11 Chen SJ, Chen GH, Chen YH, et al. Characterization of Epstein-Barrvirus miRNAome in nasopharyngeal carcinoma by deep sequencing PLoS One 2010; 5(9): e12745.,14 Jurak I, Kramer MF, Mellor JC, et al. Numerous conserved anddivergent microRNAs expressed by herpes simplex viruses 1 and 2 J Virol 2010; 84(9): 4659-72.,27 Reese TA, Xia J, Johnson LS, Zhou X, Zhang W, Virgin HW. Identification of novel microRNA-like molecules generated fromherpesvirus and host tRNA transcripts J Virol 2010; 84(19): 10344-53.] except for KSHV [10 Umbach JL, Cullen BR. In-depth analysis of Kaposi's sarcomaassociatedherpesvirus microRNA expression provides insights into themammalian microRNA-processing machinery J Virol 2010; 84(2): 695-703.]. In KSHV, two out of eleven miRNAs were found to account for 90% of the total number of reads for the viral encoded miRNA. HCMV seems to be indeed exceptional since it displays high expression of most of its miRNAs, with over 53% of the mature miRNAs having more than 10,000 reads each and with the extreme case of miR-UL22A-3p constituting 23% of the total HCMV miRNAs reads. Though the specific function of most of these miRNAs remains to be elucidated, this extremely high expression strongly suggests that they have an important biological role in the virus infection and in its interaction with the host.
Sequence abundance of known mature miRNAs, in terms of miRNA/miRNA* designation was mostly in agreement with that of the miRBase designation with a sole exception of miR-UL22A. Previous studies have reported extensive variation both in the sequences and in the relative abundance of miRBase reference sequences [10 Umbach JL, Cullen BR. In-depth analysis of Kaposi's sarcomaassociatedherpesvirus microRNA expression provides insights into themammalian microRNA-processing machinery J Virol 2010; 84(2): 695-703.,28 Amen MA, Griffiths A. Identification and expression analysis of herpesB virus-encoded small RNAs J Virol 2011; 85(14): 7296-311.-30 Umbach JL, Nagel MA, Cohrs RJ, Gilden DH, Cullen BR. Analysis ofhuman alphaherpesvirus microRNA expression in latently infectedhuman trigeminal ganglia J Virol 2009; 83(20): 10677-83.]. We noted that in contrast to other reports [31 Landgraf P, Rusu M, Sheridan R, et al. A mammalian microRNAexpression atlas based on small RNA library sequencing Cell 2007; 129(7): 1401-.] the relative expression of hcmv-miR-UL22A-3p (hcmv-miR-UL22A*) was higher (~2.6 fold higher by DS or equal to by qPCR) than that of hcmv-miR-UL22A-5p, which was the most abundant form in the miRBase.
Our DS did not reveal any reads for the mature miRNAs of mir-UL70-1 and mir-UL148D. In addition, all the reads that were mapped to miR-US4-5p were mapped with a shift of 5 nts in the 5' end to the miRBase sequence. The absence of reads of mature miRNAs of mir-UL70 and the shift in miR-US4-5p were also reported by Stark et al. [25 Stark TJ, Arnold JD, Spector DH, Yeo GW. High-resolution profilingand analysis of viral and host small RNAs during humancytomegalovirus infection J Virol 2011 Oct 19; ].
Generally, sequences of miRNAs are poorly conserved across the herpesvirus family except in their genomic localization [32 Gottwein E, Cullen BR. Viral and cellular microRNAs as determinantsof viral pathogenesis and immunity Cell Host Microbe 2008; 3(6): 375-87.-34 Cai X, Schafer A, Lu S. Epstein-Barr virus microRNAs areevolutionarily conserved and differentially expressed PLoS Pathog 2006; 2(3): e23.]. Nevertheless, whenever available, sequence conservation has an important implication in the identification and in strengthening the authenticity of new miRNAs [18 Ambros V, Bartel B, Bartel DP, et al. A uniform system for microRNAannotation RNA 2003; 9(3): 277-9.]. To this end, we investigated sequence conservation between the new miRNAs of HCMV and the CCMV genome. In our data, sequence homology between new HCMV miRNAs and predicted CCMV ranges from 67% to 100% (Table 4) with the exception of mir-UL59 and mir-UL69 which are not conserved in CCMV. It is worth noting, as was described by others [2 Dunn W, Trang P, Zhong Q, Yang E, van Belle C, Liu F. Humancytomegalovirus expresses novel microRNAs during productive viralinfection Cell Microbiol 2005; 7(11): 1684-95.,3 Grey F, Antoniewicz A, Allen E, et al. Identification andcharacterization of human cytomegalovirus-encoded microRNAs J Virol 2005; 79(18): 12095-9.,34 Cai X, Schafer A, Lu S. Epstein-Barr virus microRNAs areevolutionarily conserved and differentially expressed PLoS Pathog 2006; 2(3): e23.], that sequence homology is better conserved in mature miRNAs than in the predicted pre-miRNAs and is even more conserved at the 5' end of the mature miRNAs. This has an important implication for evolutionarily conserved function. However, such a conclusion should be taken with caution since miRNAs encoded by CCMV have not been experimentally validated yet.
miRNA offset RNAs (moRs), are 19-22 nt long fragments of RNA adjacent to an miRNA site but distant from the loop of a hairpin, have been increasingly reported recently, with the growing usage of the DS technique for in-depth analysis of miRNAs. Since its first report in Ciona intestinalis [12 Shi W, Hendrix D, Levine M, Haley B. A distinct class of small RNAsarises from pre-miRNA-proximal regions in a simple chordate NatStruct Mol Biol 2009; 16(2): 183-9.], several moRs have been identified in different organisms including mammals [13 Langenberger D, Bermudez-Santana C, Hertel J, Hoffmann S, Khaitovich P, Stadler PF. Evidence for human microRNA-offset RNAsin small RNA sequencing data Bioinformatics 2009; 25(18): 2298-301.,35 Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse ES cellsexpress endogenous shRNAs, siRNAs, and other Microprocessor-independent,Dicer-dependent small RNAs Genes Dev 2008; 22(20): 2773-85.], Drosophila [36 Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC. Evolution, biogenesis, expression, and target predictions of asubstantially expanded set of Drosophila microRNAs Genome Res 2007; 17(12): 1850-64.] and even herpes viruses [10 Umbach JL, Cullen BR. In-depth analysis of Kaposi's sarcomaassociatedherpesvirus microRNA expression provides insights into themammalian microRNA-processing machinery J Virol 2010; 84(2): 695-703.,14 Jurak I, Kramer MF, Mellor JC, et al. Numerous conserved anddivergent microRNAs expressed by herpes simplex viruses 1 and 2 J Virol 2010; 84(9): 4659-72.]. Experimental validation of the biogenesis of these small RNAs remains unclear although it has been speculated that they arise from Drosha processing of pri-miRNA, at least when expressed from a hairpin arm cleaved by Drosha [35 Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse ES cellsexpress endogenous shRNAs, siRNAs, and other Microprocessor-independent,Dicer-dependent small RNAs Genes Dev 2008; 22(20): 2773-85.]. In our current study, we also analyzed the presence of these small RNAs and identified 11 new moRs from nine hairpin-precursors. The three most abundant moRs were further confirmed by qPCR. Most of these RNAs were expressed at relatively low levels; with the following exceptions: moR-US4-5p with 1173 reads, which is barely higher than miR-US4-3p with 1145 reads and, moR-US5-2-5p with 849 reads, versus miR-US5-2-5p with 733 reads. The majority (8 out of 11) of the moRs found in our sample are derived from the 5’-side of the stem similarly to the finding reported in [13 Langenberger D, Bermudez-Santana C, Hertel J, Hoffmann S, Khaitovich P, Stadler PF. Evidence for human microRNA-offset RNAsin small RNA sequencing data Bioinformatics 2009; 25(18): 2298-301.,16 Taft RJ, Simons C, Nahkuri S, et al. Nuclear-localized tiny RNAs areassociated with transcription initiation and splice sites in metazoans Nat Struct Mol Biol 2010; 17(8): 1030-4.].
Furthermore, there is little correlation between expression levels of moRs and associated miRNAs (see Fig. 5 and Tables 2 and 3) suggesting that these RNAs are not a by-product of Drosha processing. Moreover, the fact that moRs are consistently expressed in authentic infection and the sequences of the reads had the same length and position on the precursor, does suggest that moRs might also have important biological functions.
In conclusion, we have demonstrated that by using an integrated bioinformatic/biological approach that relies heavily on DS for the analysis of HCMV transcriptome, we were able to find several new miRNAs and moRs that were not found previously. This approach has also revealed an extremely high expression of some of the mature viral encoded miRNAs which strongly suggests that they have a central biological role which remains to be elucidated.
ACKNOWLEDGEMENT
Declared none.
CONFLICT OF INTEREST
Declared none.