- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




The Open Virology Journal
(Discontinued)
ISSN: 1874-3579 ― Volume 15, 2021
Genome Stability of Pandemic Influenza A (H1N1) 2009 Based on Analysis of Hemagglutinin and Neuraminidase Genes
Emilio E Espínola*
Abstract
Influenza A virus (H1N1), which arose in 2009, constituted the fourth pandemic after the cases of 1918, 1957, and 1968. This new variant was formed by a triple reassortment, with genomic segments from swine, avian, and human influenza origins. The objective of this study was to analyze sequences of hemagglutinin (n=2038) and neuraminidase (n=1273) genes, in order to assess the extent of diversity among circulating 2009-2010 strains, estimate if these genes evolved through positive, negative, or neutral selection models of evolution during the pandemic phase, and analyze the worldwide percentage of detection of important amino acid mutations that could enhance the viral performance, such as transmissibility or resistance to drugs. A continuous surveillance by public health authorities will be critical to monitor the appearance of new influenza variants, especially in animal reservoirs such as swine and birds, in order to prevent the potential animal-human transmission of viruses with pandemic potential.
Article Information
Identifiers and Pagination:
Year: 2012Volume: 6
First Page: 59
Last Page: 63
Publisher Id: TOVJ-6-59
DOI: 10.2174/1874357901206010059
Article History:
Received Date: 18/1/2012Revision Received Date: 29/2/2012
Acceptance Date: 2/3/2012
Electronic publication date: 26/4/2012
Collection year: 2012
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Rio de la Plata y Lagerenza, CP 1120 Asunción, Paraguay; Tel: +595 21 424 520; Fax: +595 21 480 185; E-mail: emilioespinola@hotmail.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 18-1-2012 |
Original Manuscript | Genome Stability of Pandemic Influenza A (H1N1) 2009 Based on Analysis of Hemagglutinin and Neuraminidase Genes | |
INTRODUCTION
Influenza A viruses belong to the Orthomyxoviridae family, and have a genome composed of eight segments of single-stranded, negative-sense RNA. Their surfaces are composed by a lipid envelope, originated from the plasmatic membrane of infected epithelial cells, and two antigenic proteins: Hemagglutinin (HA) and Neuraminidase (NA); these two antigens exhibit higher variability compared with their remaining proteins [1 Lamb RA, Krug RM. Orthomyxoviridae: The viruses and their replication In: Knipe DM, Howley PM, Griffin DE, Eds. Fields Virology. Philadelphia: Lippincott: Williams & Wilkins 2001; pp. 1487-531.]. Depending on the extent of variability of two surface proteins, until now are known 16 HA (H1-H16), and 9 NA genotypes (N1-N9), respectively, which can be combined in different combinations [1 Lamb RA, Krug RM. Orthomyxoviridae: The viruses and their replication In: Knipe DM, Howley PM, Griffin DE, Eds. Fields Virology. Philadelphia: Lippincott: Williams & Wilkins 2001; pp. 1487-531., 2 Neumann G, Noda T, Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus Nature 2009; 459: 931-.].
In early April 2009, authorities from the Mexican public health observed a high number of influenza-like illnesses in their territory, and informed about this outbreak to the regional office of the World Health Organization (WHO). In mid April, the Centers for Disease Control from USA identified the new virus in two cases from California. The new virus spread rapidly throughout the world, and as a consequence the WHO authorities declared the “Pandemic (H1N1) 2009” on June 11, 2009 [3WHO. Pandemic (H1N1) 2009, update 75. Accessed on: January 1, 2012. Available from http://www.who.int ]. It is thought that the new 2009 H1N1 pandemic virus (from here, 2009 H1N1pdm) has emerged through at least four reassortment and transmission events among swine, avian and human H1N1 lineages, probably in Asia and North America [4 Qu Y, Zhang R, Cui P, Song G, Duan Z, Lei F. Evolutionary genomics of the pandemic 2009 H1N1 influenza viruses (pH1N 1v) Virol J 2011; 8: 250.]. Particularly, the HA segment of 2009 H1N1pdm was originated from American swine lineage, whereas the NA segment derived from the European swine lineage [5 Babakir-Mina M, Dimonte S, Perno CF, Ciotti M. Origin of the 2009 Mexico influenza virus a comparative phylogenetic analysis of the principal external antigens and matrix protein Arch Virol 2009; 154: 1349-52., 6 Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans Science 2009; 325: 197-201.]. It is believed that the ancestors of this pandemic strain remained undetected for approximately one decade due to lack of a surveillance system in pigs, the historical “mixing vessel” for new influenza viruses. Furthermore, the closest ancestors of the new pandemic strains emerged probably in January 2009 [4 Qu Y, Zhang R, Cui P, Song G, Duan Z, Lei F. Evolutionary genomics of the pandemic 2009 H1N1 influenza viruses (pH1N 1v) Virol J 2011; 8: 250.].
The objective of this study was to analyze a dataset of complete nucleotide (nt) sequences of HA and NA genes, in order to assess the extent of diversity among circulating 2009-2010 strains, estimate if these genes evolved through positive, negative, or neutral selection models of evolution during the pandemic phase, and analyze the worldwide percentage of detection of important amino acid mutations that could enhance the viral performance, such as transmissibility or resistance to drugs.
Complete CoDing Sequences (CDS) of HA (1701 nt) and NA (1410 nt) genes corresponding to 2009 H1N1pdm, isolated from humans, were downloaded from the Influenza Virus Resource (http://www.ncbi.nlm.nih.gov/genomes/FLU /SwineFlu.html) from the National Center for Biotechnology Information, by the year of sequence repository. The first dataset consisted of 3765 HA and 2996 NA sequences, respectively, which were reported in the period 2009-2010. After discarding exact duplicates in sequence using a Perl script, we obtained 2038 HA and 1273 NA sequences, respectively; these sequences were different in at least one nucleotide among all representatives. Reassortant strains were discarded, as well as incomplete CDS sequences.
Nucleotide sequences were manually edited in FASTA format, using BioEdit v7.0.5 [7 Hall TA. BioEdit a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT Nucl Acids Symp Ser 1999; 41: 95-8.], and aligned with CLUSTAL W [8 Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice Nucleic Acids Res 1994; 22: 4673-80.]. Sequence information (GenBank accession number, strain, and year of isolation) for each sample used in this study are available for HA (Table S1) and NA genes (Table S2), respectively. Pairwise distances were calculated with MEGA v5 [9 Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5 molecular evolutionary genetics analysis using maximum likelihood evolutionary distance and maximum parsimony methods Mol Biol Evol 2011. Epub ahead of print]. The percentages of identities were calculated by applying the formula 100 - (pairwise distance value x 100). A graph was constructed by plotting the percentage identities in the abscissa (x axis) vs the frequency of each of the calculated pairwise identities in the ordinate (y axis). The graphs were prepared in the R environment, using ggplot2 package (www.r-project.org).
The models of nucleotide substitution that best fitted each dataset were determined with MEGA v5 [9 Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5 molecular evolutionary genetics analysis using maximum likelihood evolutionary distance and maximum parsimony methods Mol Biol Evol 2011. Epub ahead of print], and were: GTR+I model for HA genes, and T92+G model for NA genes, respectively. Phylogenetic relationships were reconstructed by the Neighbor-Joining method [10 Saitou N, Nei M. The neighbor-joining method a new method for reconstructing phylogenetic trees Mol Biol Evol 1987; 4: 406-25.], with the appropriate models of nucleotide substitution for each dataset (as described above) and bootstrap analysis of 1000 replicates, as incorporated in MEGA v5 [9 Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5 molecular evolutionary genetics analysis using maximum likelihood evolutionary distance and maximum parsimony methods Mol Biol Evol 2011. Epub ahead of print]. Outgroup sequences for HA and NA genes corresponded to strain A/Puerto Rico/8/1934.
Mutations in each CDS were analyzed by the method of Nei and Gojobori [11 Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions Mol Biol Evol 1986; 3: 418-26.]. Codon aligned sequences for each dataset were analyzed using the Perl-based SNAP program (http://www.hiv.lanl.gov/content/sequence/SNAP/SNAP.html) [12 Korber B. HIV Signature and Sequence Variation Analysis Computational Analysis of HIV Molecular Sequences. Dordrecht, Netherlands: Kluwer Academic Publishers 2000.] in order to calculate the variability of each CDS. The selective pressure was measured by comparing the rate of non-synonymous nucleotide substitutions per non-synonymous site (dN) against that of synonymous substitutions per synonymous site (dS). The ratio dN/dS was used as an index to assess positive selection. A ratio dN/dS >1 means positive (diversifying) selection, =1 means neutral selection, and <1 means negative (purifying) selection.
The analysis of pairwise identity frequencies showed high percentage of similarities among circulating 2009-2010 pandemic influenza strains (Fig. 1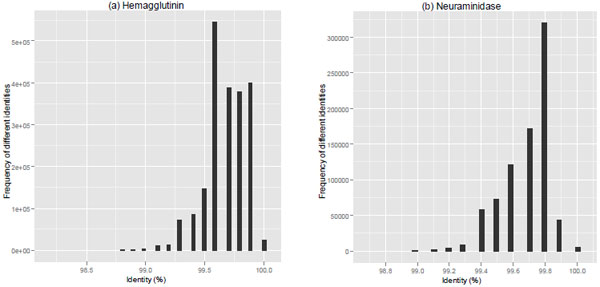 ). The average percentage of identity was 99.7% for both HA and NA genes. Thus, in this period of pandemic circulation, both genes did not segregate into different clusters, but on the contrary showed a constant and stable evolution.
). The average percentage of identity was 99.7% for both HA and NA genes. Thus, in this period of pandemic circulation, both genes did not segregate into different clusters, but on the contrary showed a constant and stable evolution.
The high percentages of nucleotide identity were in accordance with the single clustering of all 2009-2010 strains in the phylogenetic tree of HA and NA genes (Fig. 2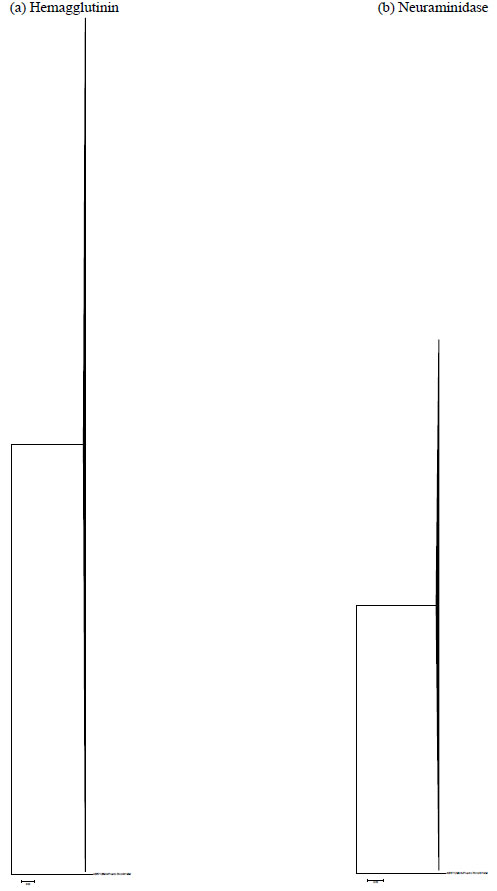 ), without temporal or geographical distribution. It is interesting to note that the overall genetic diversity among 2009 H1N1pdm was less than typically observed among seasonal influenza. This is in accordance with its short period of time of circulation in humans [13 Galiano M, Agapow PM, Thompson C, et al. Evolutionary
pathways of the pandemic influenza A (H1N1) 2009 in the UK
PLoS One 2011; 6: e23779.]. The single clustering of 2009 H1N1pdm observed in this report, however, is in contrast with other studies [14 Fereidouni SR, Beer M, Vahlenkamp T, Starick E. Differentiation
of two distinct clusters among currently circulating influenza
A(H1N1)v viruses, March-September 2009 Euro Surveill 2009; 14pii: 19409], in which the authors observe differences by using small datasets of sequences. The single clustering of 2009 H1N1pdm, furthermore, agrees with serological data in which it was observed that antigenically, the new pandemic viruses were all similar [6 Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic
characteristics of swine-origin 2009 A(H1N1) influenza viruses
circulating in humans Science 2009; 325: 197-201.], and thus not requiring a new update of the vaccine (strain A/California/07/2009) until now.
), without temporal or geographical distribution. It is interesting to note that the overall genetic diversity among 2009 H1N1pdm was less than typically observed among seasonal influenza. This is in accordance with its short period of time of circulation in humans [13 Galiano M, Agapow PM, Thompson C, et al. Evolutionary
pathways of the pandemic influenza A (H1N1) 2009 in the UK
PLoS One 2011; 6: e23779.]. The single clustering of 2009 H1N1pdm observed in this report, however, is in contrast with other studies [14 Fereidouni SR, Beer M, Vahlenkamp T, Starick E. Differentiation
of two distinct clusters among currently circulating influenza
A(H1N1)v viruses, March-September 2009 Euro Surveill 2009; 14pii: 19409], in which the authors observe differences by using small datasets of sequences. The single clustering of 2009 H1N1pdm, furthermore, agrees with serological data in which it was observed that antigenically, the new pandemic viruses were all similar [6 Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic
characteristics of swine-origin 2009 A(H1N1) influenza viruses
circulating in humans Science 2009; 325: 197-201.], and thus not requiring a new update of the vaccine (strain A/California/07/2009) until now.
Given that 2009 H1N1pdm constituted a homogeneous phylogenetic group, it was hypothesized that the diversity in nucleotide sequences localized (in average) in the 0.3% of differences within each analyzed gene. Taking into account the complete CDS for HA and NA genes, this percentage of differences constitutes approximately four to five nucleotide random variations among circulating strains.
Calculation of average dN/dS rates of evolution showed that both HA and NA genes evolved through negative (purifying) selection (Table 1), with dN/dS values of 0.2762 and 0.1939, respectively. Even though in general, both genes underwent negative selection, some positions can evolve through positive selection. For example, an early study showed that two sites involved in receptor binding specificity of HA (220 and 278) were under positive selection, and these sites were not found in swine or seasonal H1N1 viruses [15 Furuse Y, Shimabukuro K, Odagiri T, et al. Comparison of selection pressures on the HA gene of pandemic (2009) and seasonal human and swine influenza A H1 subtype viruses Virology 2010; 405: 314-21.]. Thus, changes in receptor binding sites could lead to alterations in receptor binding specificities.
In other viruses such as SARS-CoV, it was observed that they can develop through positive selection through the cross-species transmission in early epidemics, and negative selection during late epidemics [16The Chinese SARS Molecular Epidemiology Consotium. Evolution of the SARS coronavirus during the course of the SARS epidemic in China Science 2004; 303: 1666-9.]. It is possible that the same mechanism was the driven force of evolution of 2009 H1N1pdm, with positive selection at least during cross-species transmission.
A number of different amino acid mutations that could confer new functionalities to the new virus were reported worldwide, including those related to increased pathogenicity or antiviral resistance (Table 2).
Polymorphism at position 239 in HA has been associated with severe clinical outcomes, especially in immunocompromised patients; in particular, substitution 239G was found to correlate with fatal outcomes in different countries [17 Ferreira JL, Borborema SE, Brigido LF, de Oliveira MI, de Paiva TM, dos Santos CL. Sequence analysis of the 2009 pandemic influenza A H1N1 virus haemagglutinin gene from 2009-2010 Brazilian clinical samples Mem Inst Oswaldo Cruz 2011; 106: 613-., 18 Kilander A, Rykkvin R, Dudman S, Hungnes O. Observed association between the HA1 mutation D222G in the 2009 pandemic influenza A(H1N1) virus and severe clinical outcome Norway 2009-2010 Euro Surveill 2010; 15pii: 19498]. Furthermore, this mutation can arise de novo from wildtype (D239) virus in the same patient throughout the disease course [19 Chan PK, Lee N, Joynt GM, et al. Clinical and virological course of infection with haemagglutinin D222G mutant strain of 2009 pandemic influenza A (H1N1) virus J Clin Virol 2011; 50: 320-4.]. Mutation at position 239 can induce alterations in the receptor binding site, and 239G mutants bind a broader range of α2-3-linked sialyl receptors sequences expressed on cells from the lower respiratory tract, which suggested that its presence could be responsible for the exacerbation of disease [20 Liu Y, Childs RA, Matrosovich T, et al. Altered receptor specificity and cell tropism of D222G hemagglutinin mutants isolated from fatal cases of pandemic A(H1N1) 2009 influenza virus J Virol 2010; 84: 12069-74.]. Mutants 239E target mainly non-ciliated cells. We found no significant difference between sequences bearing mutations 239G (2.6%) and 239E (5.5%). The low percentage of global circulation of mutants 239G found in this study is in accordance with its lower potential to transmit to other individuals [21 Puzelli S, Facchini M, Spagnolo D, et al. Transmission of hemagglutinin D222G mutant strain of pandemic (H1N1) 2009 virus Emerg Infect Dis 2010; 16: 863-5.].
Positions 239 and 220 are localized within the HA antigenic site called Ca. The amino acid S220, though not exposed to the surface, is localized in the receptor binding domain (RBD), and its change could affect the transmissibility and infectivity of H1N1 in humans. The fixed mutation, S220T, has been found at high percentage (76.7%) in this study. To test whether change 220T could contribute to antigenic drift, it would be interesting to compare its antigenic profile against a wildtype isolate (S220). This mutation, probably, has become fixed in all pandemic strains through optimization of viral fitness, rather than immune selection or adaptation to the host.
Substitution S101N has been proposed previously as a reversion to the seasonal H1N1 residue 101N and thus possibly an adaptation to the human host, being found in some studies at high frequencies. Its global impact, however, is controvertible because it was found in only 0.2% of our sequences. Substitution E391K, found at 15.6% in our study, has been identified as part of a highly conserved epitope in the 1918 H1N1 virus with a possible role in membrane fusion [22 Ekiert DC, Bhabha G, Elsliger MA, et al. Antibody recognition of a highly conserved influenza virus epitope Science 2009; 324: 246-51.]. Another proximal substitution found in other studies, N387H, was found in only 1.7% of our sequences.
In the NA gene, it was showed that mutations V106I and N248D were present in samples at increasing numbers through early pandemic month (April to December 2009) [23 Pan C, Cheung B, Tan S, et al. Genomic signature and mutation trend analysis of pandemic (H1N1) 2009 influenza A virus PLoS One 2010; 5: e9549.]. We found both mutations at high percentages, 85.1% and 85.9% respectively, in our dataset. Change 106I was present in the 20th century cases of H1N1 (in 1918 [pandemic], and 1977), as well as 248D (in 1977). Since residue at position 248 is located at the drug target domain (DTD) region, as residue 275, it could potentially affect the sensitivity to NA inhibitors. Another substitution of possible interest in NA sequences is D199N, which was previously associated with an increase in oseltamivir resistance in both seasonal and H5N1 virus strains [24 Deyde VM, Nguyen T, Bright RA, et al. Detection of molecular markers of antiviral resistance in influenza A (H5N1) viruses using a pyrosequencing method Antimicrob Agents Chemother 2009; 53: 1039-47.]. We found, however, only 4 out of 1273 NA sequences (0.3%) containing this change. The rare substitution I223R, which was reported in association with resistance to oseltamivir, zanamivir, and peramivir [25 van der Vries E, Stelma FF, Boucher CA. Emergence of a multidrug-resistant pandemic influenza A (H1N1) virus N Engl J Med 2010; 363: 1381-2.], was also found in only 2 out of 1273 NA sequences (0.2%). Substitution H275Y has been related to oseltamivir resistance, especially in immunocompromised or severely ill persons [26 Harvala H, Gunson R, Simmonds P, et al. The emergence of oseltamivir-resistant pandemic influenza A (H1N1) 2009 virus amongst hospitalised immunocompromised patients in Scotland November-December, 2009 Euro Surveill 2010; 15: 19536.]. It was found, however, in sporadic cases in most of the countries at low frequencies (~1%) [27 Longtin J, Patel S, Eshaghi A, et al. Neuraminidase-inhibitor resistance testing for pandemic influenza A (H1N1) 2009 in Ontario Canada J Clin Virol 2011; 50: 257-61.]. In our study, we found 2% of sequences containing this change.
In conclusion, the stable evolution of 2009 H1N1pdm offers an opportunity to control its spread and prevent infections. Reports about new mutations, however, will still be important if those changes can confer an enhanced transmissibility or resistance to drugs. Furthermore, a continuous surveillance by public health authorities will be critical to monitor the appearance of new influenza variants, especially in animal reservoirs such as swine and birds, in order to prevent the potential animal-human transmission of viruses with pandemic potential.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s web site along with the published article.
ACKNOWLEDGEMENT
This work (project code: INV11) was supported by the Consejo Nacional de Ciencia y Tecnología (CONACYT) ― Programa de Apoyo al Desarrollo de la Ciencia, Tecnología e Innovación en Paraguay (BID 1698/OC-PR).
CONFLICT OF INTEREST
Declared none.
REFERENCES
| [1] | Lamb RA, Krug RM. Orthomyxoviridae: The viruses and their replication In: Knipe DM, Howley PM, Griffin DE, Eds. Fields Virology. Philadelphia: Lippincott: Williams & Wilkins 2001; pp. 1487-531. |
| [2] | Neumann G, Noda T, Kawaoka Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus Nature 2009; 459: 931-. |
| [3] | WHO. Pandemic (H1N1) 2009, update 75. Accessed on: January 1, 2012. Available from http://www.who.int |
| [4] | Qu Y, Zhang R, Cui P, Song G, Duan Z, Lei F. Evolutionary genomics of the pandemic 2009 H1N1 influenza viruses (pH1N 1v) Virol J 2011; 8: 250. |
| [5] | Babakir-Mina M, Dimonte S, Perno CF, Ciotti M. Origin of the 2009 Mexico influenza virus a comparative phylogenetic analysis of the principal external antigens and matrix protein Arch Virol 2009; 154: 1349-52. |
| [6] | Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans Science 2009; 325: 197-201. |
| [7] | Hall TA. BioEdit a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT Nucl Acids Symp Ser 1999; 41: 95-8. |
| [8] | Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice Nucleic Acids Res 1994; 22: 4673-80. |
| [9] | Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5 molecular evolutionary genetics analysis using maximum likelihood evolutionary distance and maximum parsimony methods Mol Biol Evol 2011. Epub ahead of print |
| [10] | Saitou N, Nei M. The neighbor-joining method a new method for reconstructing phylogenetic trees Mol Biol Evol 1987; 4: 406-25. |
| [11] | Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions Mol Biol Evol 1986; 3: 418-26. |
| [12] | Korber B. HIV Signature and Sequence Variation Analysis Computational Analysis of HIV Molecular Sequences. Dordrecht, Netherlands: Kluwer Academic Publishers 2000. |
| [13] | Galiano M, Agapow PM, Thompson C, et al. Evolutionary pathways of the pandemic influenza A (H1N1) 2009 in the UK PLoS One 2011; 6: e23779. |
| [14] | Fereidouni SR, Beer M, Vahlenkamp T, Starick E. Differentiation of two distinct clusters among currently circulating influenza A(H1N1)v viruses, March-September 2009 Euro Surveill 2009; 14pii: 19409 |
| [15] | Furuse Y, Shimabukuro K, Odagiri T, et al. Comparison of selection pressures on the HA gene of pandemic (2009) and seasonal human and swine influenza A H1 subtype viruses Virology 2010; 405: 314-21. |
| [16] | The Chinese SARS Molecular Epidemiology Consotium. Evolution of the SARS coronavirus during the course of the SARS epidemic in China Science 2004; 303: 1666-9. |
| [17] | Ferreira JL, Borborema SE, Brigido LF, de Oliveira MI, de Paiva TM, dos Santos CL. Sequence analysis of the 2009 pandemic influenza A H1N1 virus haemagglutinin gene from 2009-2010 Brazilian clinical samples Mem Inst Oswaldo Cruz 2011; 106: 613-. |
| [18] | Kilander A, Rykkvin R, Dudman S, Hungnes O. Observed association between the HA1 mutation D222G in the 2009 pandemic influenza A(H1N1) virus and severe clinical outcome Norway 2009-2010 Euro Surveill 2010; 15pii: 19498 |
| [19] | Chan PK, Lee N, Joynt GM, et al. Clinical and virological course of infection with haemagglutinin D222G mutant strain of 2009 pandemic influenza A (H1N1) virus J Clin Virol 2011; 50: 320-4. |
| [20] | Liu Y, Childs RA, Matrosovich T, et al. Altered receptor specificity and cell tropism of D222G hemagglutinin mutants isolated from fatal cases of pandemic A(H1N1) 2009 influenza virus J Virol 2010; 84: 12069-74. |
| [21] | Puzelli S, Facchini M, Spagnolo D, et al. Transmission of hemagglutinin D222G mutant strain of pandemic (H1N1) 2009 virus Emerg Infect Dis 2010; 16: 863-5. |
| [22] | Ekiert DC, Bhabha G, Elsliger MA, et al. Antibody recognition of a highly conserved influenza virus epitope Science 2009; 324: 246-51. |
| [23] | Pan C, Cheung B, Tan S, et al. Genomic signature and mutation trend analysis of pandemic (H1N1) 2009 influenza A virus PLoS One 2010; 5: e9549. |
| [24] | Deyde VM, Nguyen T, Bright RA, et al. Detection of molecular markers of antiviral resistance in influenza A (H5N1) viruses using a pyrosequencing method Antimicrob Agents Chemother 2009; 53: 1039-47. |
| [25] | van der Vries E, Stelma FF, Boucher CA. Emergence of a multidrug-resistant pandemic influenza A (H1N1) virus N Engl J Med 2010; 363: 1381-2. |
| [26] | Harvala H, Gunson R, Simmonds P, et al. The emergence of oseltamivir-resistant pandemic influenza A (H1N1) 2009 virus amongst hospitalised immunocompromised patients in Scotland November-December, 2009 Euro Surveill 2010; 15: 19536. |
| [27] | Longtin J, Patel S, Eshaghi A, et al. Neuraminidase-inhibitor resistance testing for pandemic influenza A (H1N1) 2009 in Ontario Canada J Clin Virol 2011; 50: 257-61. |