- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




The Open Virology Journal
(Discontinued)
ISSN: 1874-3579 ― Volume 15, 2021
Both Cyclophilin Inhibitors and Direct-Acting Antivirals Prevent PKR Activation in HCV-Infected Cells
Michael Bobardt, Udayan Chatterji, Precious Lim, Katarzyna Gawlik, Philippe Gallay*
Abstract
We and others demonstrated that the contact between NS5A and the host factor CypA is critical for HCV replication. CypI, by disrupting NS5A-CypA complexes, block HCV replication both in vitro and in patients. Since NS5A also binds to PKR, a central component of the IFN response, we investigated the possibility of a relationship between CypA, NS5A and PKR in the IFN response to HCV. HCV-infected cells treated with CypI, DAAs or IFN were analyzed for the expression and activation of various components of the innate response. We found that CypI (cyclosporine A, alisporivir, NIM811 and sanglifehrins), drastically prevented the activation/phosphorylation, but not the expression of IFN-induced PKR in HCV-infected cells. CypI had no effect on the expression or phosphorylation of other components of the innate response such as eiF2, NF-kB, IRF3, IRF9, STAT1 and STAT2, suggesting a specific effect on PKR. No significant activation of IFN-induced PKR was observed in the absence of HCV. Importantly, we found that several classes of DAAs such as NS3/4A protease, NS5B polymerase and NS5A inhibitors also prevented PKR activation. Furthermore, we found that PKR activation by the dsRNA mimic poly I:C cannot be prevented by CypI or DAAs. Our findings suggest that CypI do not have a unique effect on PKR activation, but rather the suppression of HCV replication by any anti-HCV inhibitor, abrogates PKR activation induced by IFN. Moreover, they suggest that the accumulation of dsRNA intermediates allows HCV to exploit the activation of PKR to counteract the IFN response.
Article Information
Identifiers and Pagination:
Year: 2014Volume: 8
First Page: 1
Last Page: 8
Publisher Id: TOVJ-8-1
DOI: 10.2174/1874357901408010001
Article History:
Received Date: 5/1/2014Revision Received Date: 6/3/2014
Acceptance Date: 7/3/2014
Electronic publication date: 7/3/2014
Collection year: 2014
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Department of Immunology & Microbial Science, IMM-9, The Scripps Research Institute, 10550 N. Torrey Pines Rd., La Jolla, CA 92037, USA; Tel: (858) 784-8180; Fax: (858) 784-8831; E-mail: gallay@scripps.edu
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 5-1-2014 |
Original Manuscript | Both Cyclophilin Inhibitors and Direct-Acting Antivirals Prevent PKR Activation in HCV-Infected Cells | |
INTRODUCTION
Chronic hepatitis C affects approximately 200 million people worldwide and is a leading cause of acute and chronic liver diseases [1Dienstag JL, McHutchison JG. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology quiz 214-237 2006; 130: 231-64.] and 4 million new HCV infections occur each year [2Alter MJ. Epidemiology of hepatitis C virus infection. World J Gastroenterol 2007; 13: 2436-41., 3Soriano V, Madejon A, Vispo E , et al. Emerging drugs for hepatitis C. Expert Opin Emerg Drugs 2008; 13: 1-19.]. HCV accounts for two third of liver cancer and transplant cases in the developed world [4Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 2005; 5: 558-67.], and 12,000-15,000 individuals are projected to die from HCV every year in the U.S. [5Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States. 1999 through 2002. Ann Intern Med 2006; 144: 705-14.]. Until 2011, the combination of pegylated IFNα and RBV had a success rate of ~80% in patients with GT2 and GT3, but only ~50% in patients with GT1 and most importantly causes severe side effects [6Cross TJ, Antoniades CG, Harrison PM. Current and future management of chronic hepatitis C infection. Postgrad Med J 2008; 84: 172-6.-10Tong MJ, Reddy KR, Lee WM , et al. Treatment of chronic hepatitis C with consensus interferon: a multicenter. randomzed.controlled trial. Consensus Interferon Study Group. Hepatology 1997; 26: 747-54.]. There is thus an imperative demand for the identification and development of supplementary anti-HCV agents with diversified mechanisms of action in order to deliver interchangeable therapies for patients who are poorly responsive or non-responsive to the pegylated IFNα/RBV standard of care [1Dienstag JL, McHutchison JG. American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology quiz 214-237 2006; 130: 231-64., 2Alter MJ. Epidemiology of hepatitis C virus infection. World J Gastroenterol 2007; 13: 2436-41.]. A novel class of anti-HCV agents has recently emerged: the HTAs cyclophilin inhibitors [11Fischer G, Gallay P, Hopkins S. Cyclophilin inhibitors for the treatment of HCV infection. Curr Opin Investig Drugs 2010; 11: 911-8.-19Gallay PA, Lin K. Profile of alisporivir and its potential in the treatment of hepatitis C. Drug Des Dev Ther 2013; 7: 105-5.]. Three cyclophilin inhibitors – alisporivir, NIM811 and SCY-635 have been tested in early phase I and II studies [20Flisiak R, Horban A, Gallay P , et al. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 2008; 47: 817-26.-30Griffel L, Bao W, Orsenigo R , et al. Interferon (IFN)-free alisporivir has a better overall safety profile compared to IFN-containing treatment: a pooled analysis of the ALV development program. J Hepatol 2013; 58 (Suppl 1 ): 336-7.]. Alisporivir is the most advanced in this class, with the largest number of patients exposed and currently is in clinical development as IFN-free treatment.
We and others obtained evidence that HCV relies on host CypA to replicate in human hepatoma cells [31Liu Z, Yang F, Robotham JM, Tang H. Critical role of cyclophilin A and its prolyl-peptidyl isomerase activity in the structure and function of the hepatitis C virus replication complex. J Virol 2009; 83: 6554-.-33Chatterji U, Bobardt MD, Selvarajah S , et al. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J Biol Chem 2009; 284: 16998-7005.] as well as in humanized mice [34Dorner M, Horwitz JA, Donovan BM , et al. Completion of the entire hepatitis C virus life cycle in genetically humanized mice. Nature 2013; 501: 237-41.]. Since CypA represents the major intracellular target for CypI such as cyclosporine A [35Harding MW, Handschumacher RE. Cyclophilin a primary molecular target for cyclosporine. Structural and functional implications Transplantation 1988; 46(Suppl 2 ): 29-35.], it has been proposed that they block HCV replication by neutralizing the peptidyl-prolyl isomerase activity of CypA. We and others also demonstrated that CypA can interact directly with the HCV NS5A protein and that most importantly, CypI prevent NS5A-CypA interactions in a dose-dependent manner [36Hanoulle X, Badillo A, Wieruszeski JM , et al. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J Biol Chem 2009; 284: 13589-601.-39Coelmont L, Hanoulle X, Chatterji U , et al. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 2010; 5: e13687.]. We also showed that the interaction between NS5A and CypA is conserved among HCV genotypes and that CypI block all these interactions [38Chatterji U, Lim P, Bobardt MD , et al. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J Hepatol 2010; 53: 50-6.]. These in vitro data are in accordance with the fact that CypI are pangenotypic both in vitro and in patients. There is thus a direct correlation between disrupting NS5A-CypA complexes and blocking HCV replication. The Lippens and the Hanoulle labs elegantly showed that CypA induces cis-trans isomerization of several proline residues within the domains II and III of NS5A [36Hanoulle X, Badillo A, Wieruszeski JM , et al. Hepatitis C virus NS5A protein is a substrate for the peptidyl-prolyl cis/trans isomerase activity of cyclophilins A and B. J Biol Chem 2009; 284: 13589-601., 39Coelmont L, Hanoulle X, Chatterji U , et al. DEB025 (Alisporivir) inhibits hepatitis C virus replication by preventing a cyclophilin A induced cis-trans isomerisation in domain II of NS5A. PLoS One 2010; 5: e13687., 40Verdegem D, Badillo A, Wieruszeski JM , et al. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J Biol Chem 2011; 286(23): 20441-54.]. Interestingly, CypA and the NS5B polymerase share a common binding site on NS5A [41Rosnoblet C, Fritzinger B, Legrand D , et al. Hepatitis C virus NS5B and host cyclophilin A share a common binding site on NS5A. J Biol Chem 2012; 287(53): 44249-60.]. However, it remains obscure how CypA, by binding to NS5A and/or by isomerizing NS5A, potentiates HCV replication.
The IFN-inducible PKR plays multiple roles in a cell, in response to different stress situations. As a member of the ISGs, PKR was recognized as a factor in the antiviral action of IFN [42Samuel CE. Antiviral actions of interferon.Interferon-regulated cellular proteins and their surprisingly selective antiviral activities. Virology 1991; 183: 1-11.], due to its ability to control translation, through phosphorylation, of the α subunit of eIF2a. As such, PKR participates in the generation of stress granules or autophagy, and a number of viruses have developed strategies to inhibit its action. Mutations within the PKR-binding region of NS5A, including those within the ISDR, disrupt NS5A-PKR interactions [43Gale M Jr, Blakely CM, Kwieciszewski B , et al. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol Cell Biol 1998; 18(9): 5208-18.]. Gale et al. showed that NS5A interacts in vitro with PKR [43Gale M Jr, Blakely CM, Kwieciszewski B , et al. Control of PKR protein kinase by hepatitis C virus nonstructural 5A protein: molecular mechanisms of kinase regulation. Mol Cell Biol 1998; 18(9): 5208-18.]. Previous studies nicely demonstrated that NS5A is an RNA binding protein [44Huang L, Hwang J, Sharma SD , et al. Hepatitis C virus nonstructural protein 5A (NS5A) is an RNA-binding protein. J Biol Chem 2005; 280: 36417-28., 45Hwang J, Huang L, Cordek DG , et al. Hepatitis C virus nonstructural protein 5A: biochemical characterization of a novel structural class of RNA-binding proteins. J Virol 2010; 84: 12480-91.], which can regulate the binding of PKR to the IRES of the HCV RNA [46Toroney R, Nallagatla SR, Boyer JA, Cameron CE, Bevilacqua PC. Regulation of PKR by HCV IRES RNA: importance of domain II and NS5A. J Mol Biol 2010; 400: 393-412.]. Based on these findings, it has been proposed that the NS5A-PKR interaction serves as a target for therapeutic strategies against HCV. Since we and others obtained several lines of evidence suggesting that the NS5A-CypA interaction also represents an attractive target for the development of anti-HCV agents such as CypI, we asked in this study whether CypA and PKR act in concert to regulate HCV replication.
MATERIALS AND METHODS
Compounds
The HCV NS5A inhibitor daclatasvir (Bristol Myers Squibb), the HCV NS5B polymerase inhibitor sofosbuvir (Gilead), the HCV NS3 protease inhibitors boceprevir (Merck) and telaprevir (Vertex) and the HIV-1 reverse transcriptase inhibitor emtricitabine (Gilead) were all obtained from MedChemexpress (Princeton, NJ 08540, USA). Alisporivir and NIM811 were generously provided by Novartis, whereas cyclosporine A, sanglifehrins A and B were generously provided by Drs. Wilkinson and Gregory. Poly I:C was obtained from InvivoGen (San Diego, CA, USA).
Replicons
The GT2a subgenomic JFH-1 replicon was generously provided by Drs. T. Wakita and F. Chisari. The GT2a geno-mic luciferase reporter replicon Luc-Neo-JFH-1 was created as follows. The plasmid pFK-Luc-JFH1 was generously obtained from Drs. T. Wakita and T. Pietschmann [47Wakita T, Pietschmann T, Kato T , et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 2005; 11: 791-6., 48Koutsoudakis G, Kaul A, Steinmann E , et al. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 2006; 80: 5308-20.] and the XbaI site in the firefly luciferase gene, and the NotI site in the EMCV IRES were utilized to clone the Luci-ferase/Ubiquitin-NPT II fusion cassette out of pFK389I Luc-Neo (wild-type replicon from GT1b) (generous gift from Dr. R. Bartenschlager) [48Koutsoudakis G, Kaul A, Steinmann E , et al. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 2006; 80: 5308-20., 49Vrolijk JM, Kaul A, Hansen BE , et al. A replicon-based bioassay for the measurement of interferons in patients with chronic hepatitis C. J Virol Methods 2003; 110: 201-9.] and placed into the pFK-Luc-JFH1 plasmid, creating the full-length Luc-Neo-JFH-1 con-struct. Replicons were stably expressed in Huh7.5.1 cells under G418 selection.
Antibodies
Anti-PKR, anti-eiF2, anti-IRF3, anti-IRF9, anti-NF-kB and anti-OAS1 antibodies were obtained from Santa Cruz; the anti-phospho-PKR antibody was obtained from Abcam; anti-phospho-eiF2, anti-STAT1, anti-phospho-STAT1 antibody were obtained from Cell Signaling Technologies; the anti-NS5A antibody (9E10) was generously obtained by Dr. C. Rice; and anti-calnexin antibody was obtained from Sigma.
PKR Activation
Parental, genomic or subgenomic JFH-1-expressing Huh7.5.1 cells plated for 24 h were treated with or without CypI or direct-acting antivirals (daclatasvir, sofosbuvir, boceprevir, telaprevir and emtricitabine). Cells were then treated for 24 h with IFNα (300 U/mL) and lysed. Lysates were standardized for protein content and analyzed by Western blotting for their content in various host and viral proteins.
RESULTS
Alisporivir Prevents PKR Activation
We chose the potent non-immunosuppressive CypI alisporivir to determine the effect of CypA neutralization on PKR activation. We also chose to employ the JFH-1 cell culture system for productive HCV infection [47Wakita T, Pietschmann T, Kato T , et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 2005; 11: 791-6., 50Zhong J, Gastaminza P, Cheng G , et al. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci USA 2005; 102: 9294-., 51Lindenbach BD, Evans MJ, Syder AJ , et al. Complete replication of hepatitis C virus in cell culture. Science 2005; 309: 623-.] since it allows more direct measurements of the effects of the IFN response during the life cycle of the virus. To induce PKR activation, IFNα or IFNβ were added to HCV-infected cells pre-incubated with or without alisporivir. Twenty-four and 48 h post-IFN treatment, cells were washed and lysed. Lysates were analyzed for their content in various viral and host proteins. In accordance with the fact that HCV is sensitive to IFN in vitro, NS5A levels in JFH-1-infected cells treated with IFNα or IFNβ were significantly decreased 24 and 48 h post-IFN treatment (Fig. 1A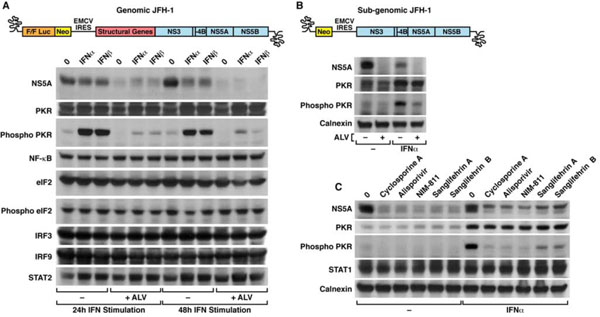 ). NS5A levels were profoundly reduced in alisporivir-treated cells compared to untreated cells (Fig. 1A), further suggesting that the CypI alisporivir, by interfering with viral RNA replication, suppresses the expression and production of HCV proteins such as NS5A. We obtained similar results for the HCV core (data not shown). We then looked at the expression of the non-phosphorylated form of PKR, also called the inactive form of PKR. A significant level of inactive/non-phosphorylated PKR pre-existed in JFH-1-infected cells and both IFNα and IFNβ increased its expression, whereas alisporivir alone had no effect (Fig. 1A). This confirms that PKR is an IFN-inducible protein and suggests that CypA neutralization does not regulate the expression of the inactive/non-phosphorylated form of PKR in an established infection. Importantly, we found that IFNα and IFNβ greatly promoted the levels of phosphorylated PKR, the active form of PKR 24 and 48 h post-IFN treatment (Fig. 1A). Remark-ably, alisporivir prevented the IFN-mediated activation/phos-phorylation of PKR (Fig. 1A). The IFN induction as well as the alisporivir-mediated inhibition of PKR activation appears to be specific for components of the innate response such as NF-κB, eIF2, its phosphorylated form (phospho-eiF2), IRF3 and IRF9 as well as STAT2 were not altered by alisporivir (Fig. 1A). Altogether these results demonstrate that CypA inhibition by alisporivir markedly decreases the IFN-induced activation/phosphorylation of PKR. These data provide the first link between HCV, CypA and PKR.
). NS5A levels were profoundly reduced in alisporivir-treated cells compared to untreated cells (Fig. 1A), further suggesting that the CypI alisporivir, by interfering with viral RNA replication, suppresses the expression and production of HCV proteins such as NS5A. We obtained similar results for the HCV core (data not shown). We then looked at the expression of the non-phosphorylated form of PKR, also called the inactive form of PKR. A significant level of inactive/non-phosphorylated PKR pre-existed in JFH-1-infected cells and both IFNα and IFNβ increased its expression, whereas alisporivir alone had no effect (Fig. 1A). This confirms that PKR is an IFN-inducible protein and suggests that CypA neutralization does not regulate the expression of the inactive/non-phosphorylated form of PKR in an established infection. Importantly, we found that IFNα and IFNβ greatly promoted the levels of phosphorylated PKR, the active form of PKR 24 and 48 h post-IFN treatment (Fig. 1A). Remark-ably, alisporivir prevented the IFN-mediated activation/phos-phorylation of PKR (Fig. 1A). The IFN induction as well as the alisporivir-mediated inhibition of PKR activation appears to be specific for components of the innate response such as NF-κB, eIF2, its phosphorylated form (phospho-eiF2), IRF3 and IRF9 as well as STAT2 were not altered by alisporivir (Fig. 1A). Altogether these results demonstrate that CypA inhibition by alisporivir markedly decreases the IFN-induced activation/phosphorylation of PKR. These data provide the first link between HCV, CypA and PKR.




We then asked whether similar results can be observed using a subgenomic JFH-1 replicon. Similarly to the infectious genomic HCV replicon, we found that IFN up-regulates the expression of PKR in cells replicating the subgenomic replicon (Fig. 1B). As for the genomic HCV replicon, we found that alisporivir prevents the IFN-induced activation/phosphorylation of PKR, has no effect of the expression of inactive/non-phosphorylated PKR and decreases the expression of HCV proteins such as NS5A (Fig. 1B). We obtained similar results for subgenomic replicons from other genotypes (1b, 3a and 4a) (data not shown). These data indicate that HCV structural proteins are not necessary for the activation of IFN-induced PKR by IFN.
We also examined whether the alisporivir impact on the inhibition of PKR activation is IFN concentration-dependent.
HCV-infected cells were treated with increasing concentrate-ions of IFNα (from 100 to 2700 U/mL). We found that a low concentration of IFNα (100 U/mL) sufficed to increase the expression of inactive/non-phosphorylated PKR, whereas an intermediate concentration of IFNα (300 U/mL) gave the most profound activation of PKR (data not shown). Alisporivir reduced the IFN-mediated activation of PKR independently of the IFN dose (data not shown). We obtained similar results for IFNβ (type I), IFNα (type II) and IFNβ (type III) (data not shown), suggesting that alisporivir prevents the activation of PKR independently of the type of IFN used for induction.
Any CypI Prevents the Activation of IFN-Induced PKR
We showed above that the treatment of HCV-infected cells with the CypI alisporivir suppresses PKR activation/ phosphorylation. We then asked whether this inhibition is specific to alisporivir or is mediated by any CypI. There are two main classes of CypI - i) cyclosporine A and derivates and ii) sanglifehrins and derivates – that both target the iso-merase pocket of CypA. We chose cyclosporine A, the two non-immunosuppressive cyclosporine A analogs – alispori-vir and NIM-811 – and sangliferins A and B. We employed the same experimental design as above except that we used IFNα exclusively and lysed HCV-infected cells 24 h post-IFN treatment. All CypI significantly decreased NS5A levels, but did not affect the IFN-induced expression of inactive/non-phosphorylated PKR (Fig. 1C). However, each CypI efficiently suppressed PKR activation/phospho-rylation
(Fig. 1C). These data suggest that inhibition of CypA prohibits the activation of IFN-induced PKR in HCV-infected cells.
DAAs Also Block PKR Activation
We then asked whether anti-HCV agents other than CypI such as DAAs also modulate the activation of PKR. Specifically, we tested the effect of the protease inhibitors boceprevir and telaprevir as well as the NS5A inhibitor daclatasvir. We used as positive control the CypI alisporivir and as negative control the HIV-1 reverse transcriptase inhibitor emtricitabine. All anti-HCV agents were potent. Indeed alisporivir, boceprevir, telaprevir and daclatasvir, but not the HIV-1 agent emtricitabine, suppressed the expression of HCV proteins such as NS5A (Fig. 2 ). Examination of calnexin levels showed that similar amounts of cell lysates were analyzed (Fig. 2). IFN elevates the expression of inactive/non-phosphorylated PKR and all inhibitors have no effect on it (Fig. 2). Similarly, IFN enhances the phosphorylation of STAT1 that is not influenced by the inhibitors (Fig. 2). Most importantly, we found that all anti-HCV agents - alisporivir, boceprevir, telaprevir and daclatas-vir – but not the HIV-1 inhibitor emtricitabine, prevent PKR activation/phosphorylation (Fig. 2). This finding is critical because it demonstrates that the inhibition of PKR activation is not unique to CypI, but is shared by any anti-HCV agents.
). Examination of calnexin levels showed that similar amounts of cell lysates were analyzed (Fig. 2). IFN elevates the expression of inactive/non-phosphorylated PKR and all inhibitors have no effect on it (Fig. 2). Similarly, IFN enhances the phosphorylation of STAT1 that is not influenced by the inhibitors (Fig. 2). Most importantly, we found that all anti-HCV agents - alisporivir, boceprevir, telaprevir and daclatas-vir – but not the HIV-1 inhibitor emtricitabine, prevent PKR activation/phosphorylation (Fig. 2). This finding is critical because it demonstrates that the inhibition of PKR activation is not unique to CypI, but is shared by any anti-HCV agents.
The latter finding led us to postulate that there is a direct correlation between PKR activation and viral replication. To test this hypothesis, we conducted similar experiments in
both non-infected and HCV-infected cells. We chose as anti-HCV agents – alisporivir, the NS5A inhibitor daclastavir and the NS5B polymerase inhibitor sofosbuvir. We first found that IFN enhances similarly (~10-fold) the expression of inactive/non-phosphorylated PKR in infected and non-infected cells (Fig. 3A ). In the absence of IFN, PKR levels in infected cells were slightly superior to those in non-infected cells (~2-fold), suggesting that HCV replication in Huh7.5.1 cells up-regulates PKR expression (Fig. 3A). In contrast to HCV-infected cells, we found that IFN fails to trigger PKR activation/phosphorylation in non-infected cells (Fig. 3A) (~ 12-fold difference), suggesting that the presence of the virus is required for optimal PKR activation. As above, we found that all anti-HCV agents prevent PKR activation/ phosphory-lation in infected cells (5-6-fold decrease) and have no effect on the expression of inactive/non-phospho-rylated PKR (Fig. 3A). The expression of non-phospho-rylated and phosphorylated STAT1 is similarly up-regulated by IFN in infected and non-infected cells (~13-fold increase) (Fig. 3A). Moreover, anti-HCV agents have no effect on the IFN-induced expression of non-phosphorylated and phos-phorylated STAT1 (Fig. 3A). As expected alisporivir, dacla-stavir, sofosbuvir and IFN, by inhibiting JFH-1 replication, decrease NS5A expression (Fig. 3A). Together, these findi-ngs suggest that the IFN-mediated activation occurs only for specific components of the IFN response during an esta-blished infection.
). In the absence of IFN, PKR levels in infected cells were slightly superior to those in non-infected cells (~2-fold), suggesting that HCV replication in Huh7.5.1 cells up-regulates PKR expression (Fig. 3A). In contrast to HCV-infected cells, we found that IFN fails to trigger PKR activation/phosphorylation in non-infected cells (Fig. 3A) (~ 12-fold difference), suggesting that the presence of the virus is required for optimal PKR activation. As above, we found that all anti-HCV agents prevent PKR activation/ phosphory-lation in infected cells (5-6-fold decrease) and have no effect on the expression of inactive/non-phospho-rylated PKR (Fig. 3A). The expression of non-phospho-rylated and phosphorylated STAT1 is similarly up-regulated by IFN in infected and non-infected cells (~13-fold increase) (Fig. 3A). Moreover, anti-HCV agents have no effect on the IFN-induced expression of non-phosphorylated and phos-phorylated STAT1 (Fig. 3A). As expected alisporivir, dacla-stavir, sofosbuvir and IFN, by inhibiting JFH-1 replication, decrease NS5A expression (Fig. 3A). Together, these findi-ngs suggest that the IFN-mediated activation occurs only for specific components of the IFN response during an esta-blished infection.
Suppression of HCV Replication Precludes PKR Activation
Previous work suggested that the recognition of dsRNA by PKR induces its dimerization and activation by autophosphorylation [52Gale MJr, Katze MG. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR. the interferon-induced protein kinase. Pharmacol Ther 1998; 78: 29-46.]. Moreover, Targett-Adams et al. nicely showed that the replication of the HCV genome involves the generation of dsRNA replicative intermediates [53Targett-Adams P, Boulant S, McLauchlan J. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J Virol 2008; 82: 2182-95.]. Based on these observations, we postulated that PKR activation/phosphorylation depends on the accumulation of HCV dsRNA during viral replication. In this scenario, blocking HCV replication by CypI or DAAs should suppress viral dsRNA accumulation and therefore PKR activation. To test this hypothesis, we took advantage of poly I:C, a structurally synthetic analog of dsRNA [54Lindh HF, Lindsay HL, Mayberry BR, Forbes M. Polyinosinic-cytidylic acid complex (poly I:C) and viral infections in mice. Proc Soc Exp Biol Med 1969; 132: 83-7.]. Poly I:C triggers PKR activation/phosphorylation in non-infected cells (Fig. 3B), suggesting that PKR recognizes the dsRNA mimic and that this recognition mediates its activation/ phosphorylation. The degree of PKR activation by poly I:C was less profound than that by HCV, but was nevertheless significant (Fig. 3B). In contrast to the HCV-mediated PKR activation/phosphorylation, alisporivir did not suppress the poly I:C-mediated PKR activation/phosphorylation (Fig. 3B). This is in agreement with our hypothesis of work that inhibiting HCV replication prevents viral dsRNA accumulat-ion and therefore precludes PKR activation/phosphorylation.
DISCUSSION
In this study, we investigated the possibility that CypI specifically modulate the IFN response to HCV. We first found that the CypI alisporivir prevents PKR activation/phosphorylation in IFNa-treated HCV-infected cells. The inhibitory effect was also observed using type II and III IFNs as well as subgenomic replicon cells. We found that several CypI and various DAAs including protease, polymerase and NS5A inhibitors all suppress the IFN-mediated PKR activation/phosphorylation in HCV-infected cells. Moreover, we showed that IFN does not trigger PKR activation/phosphorylation in cells lacking HCV. We also found that the dsRNA mimic poly I:C can substitute HCV for PKR activation/phosphorylation and that anti-HCV agents fail to prevent this activation. Based on these findings, we propose the following model (Fig. 4 ). IFN enhances the expression of the inactive/non-phosphorylated form of PKR. Newly expressed inactive PKR molecules recognize and bind to HCV dsRNA intermediates, which accumulate in the cytoplasm during viral replication when cells are cultured in the absence of anti-HCV agents. The binding of PKR to HCV dsRNA at the IRES [46Toroney R, Nallagatla SR, Boyer JA, Cameron CE, Bevilacqua PC. Regulation of PKR by HCV IRES RNA: importance of domain II and NS5A. J Mol Biol 2010; 400: 393-412.] mediates PKR dimerization and autophosphorylation. In the presence of anti-HCV agents, viral replication is stopped and HCV dsRNA accumulation suppressed. In the absence of HCV dsRNA, no PKR activation/phosphorylation can occur.
). IFN enhances the expression of the inactive/non-phosphorylated form of PKR. Newly expressed inactive PKR molecules recognize and bind to HCV dsRNA intermediates, which accumulate in the cytoplasm during viral replication when cells are cultured in the absence of anti-HCV agents. The binding of PKR to HCV dsRNA at the IRES [46Toroney R, Nallagatla SR, Boyer JA, Cameron CE, Bevilacqua PC. Regulation of PKR by HCV IRES RNA: importance of domain II and NS5A. J Mol Biol 2010; 400: 393-412.] mediates PKR dimerization and autophosphorylation. In the presence of anti-HCV agents, viral replication is stopped and HCV dsRNA accumulation suppressed. In the absence of HCV dsRNA, no PKR activation/phosphorylation can occur.
Our model is in accordance with previous work of Thomis and Samuel who demonstrated using a mixture of wild-type and kinase-deficient PKR (K296R) proteins that PKR can be autophosphorylated in an intermolecular manner [55Thomis DC, Samuel CE. Mechanism of interferon action: evidence for intermolecular autophosphorylation and autoactivation of the interferon-induced RNA-dependent protein kinase PKR. J Virol 1993; 67: 7695-00.]. This model is also in accordance with recent studies from the Chisari and the Meurs labs, which elegantly demonstrated that early HCV infection not only triggers PKR induction and activation, but also attenuates the induction of ISG protein expression despite normal induction of ISG mRNAs [56Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 2009; 6: 513-22., 57Arnaud N, Dabo S, Maillard P , et al. Hepatitis C virus controls interferon production through PKR activation. PLoS One 2010; 5(5): e10575.]. Moreover, they showed that when PKR expression is down-regulated by shRNA in IFN-treated infected cells, ISG protein induction is restored to normal levels and IFN antiviral effect is enhanced. Our findings, which are in accordance with those observations, further suggest that HCV exploits PKR activation to counteract the IFN response by preferentially suppressing the translation of ISGs, at least in the JFH-1 cell culture infection system.
Watashi et al. presented their recent findings both at the 2013 European Association for the Study of the Liver (EASL) Congress (Amsterdam, Netherlands) and the 2013 20th International Symposium on HCV and Related Viruses (Melbourne, Australia), suggesting a novel regulation mechanism of IFN signaling by CypA in HCV-infected cells [58Watashi K, Daito T, Sluder A, Borroto-Esoda K, Wakita T. Cyclophilin inhibitors potentiate interferon signaling through diminished PKR phosphorylation in HCV-infected cells. 48th Annual Meeting of the European Association for the Study of the Liver 2013; 58: S632., 59Watashi K, Daito T, Sluder A, Nakajima S, Borroto-Esoda K, Wakita T. International Symposium on Hepatitis C Virus and Related Viruses. Melbourne 2013 Novel regulation mechanism of interferon signaling by cyclophilin through modulation of PKR in HCV-infected cells Abstract 32218 ]. They confirmed published results from the Chisari and the Meurs labs showing that ISG protein expression induction is attenuated during HCV infection [56Garaigorta U, Chisari FV. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 2009; 6: 513-22., 57Arnaud N, Dabo S, Maillard P , et al. Hepatitis C virus controls interferon production through PKR activation. PLoS One 2010; 5(5): e10575.]. Interestingly, Watashi et al. found that the CypI SCY-635, but not the protease inhibitor telaprevir, restores the IFN-mediated up-regulation of ISG proteins and prevents PKR activation/phosphorylation. Since the effect of telaprevir was different from that of the CypI SCY-635, they concluded that the restoration of ISG protein up-regulation as well as the inhibition of PKR activation do not result from the sup-pression of HCV replication. Based on these findings, they proposed that CypI exert two distinct anti-HCV activities: an antiviral activity that suppresses viral replication directly, i.e., rupture of NS5A-CypA interactions, and another that inhibits viral replication indirectly, via the IFN signaling. Our present results are apparently conflicting since we convincingly demonstrated that any anti-HCV agents - CypI, protease (telaprevir and boceprevir), polymerase (sofosbuvir) and NS5A (daclatasvir) inhibitors – all prevent PKR activation and suppress HCV replication. We originally thought that this discrepancy originated from the use of different Huh7 cell lines since Watashi et al. used Huh7 cells and that the data presented here were generated using Huh7.5.1 cells. However, we conducted similar experiments with Huh7.5 and Huh7 cells and obtained comparable results (data not shown to avoid redundancy). The main reason why we present only data obtained using Huh7.5.1 cells created by the Chisari lab (49) is because this cell line is uniquely design to recapitulate all HCV infection and replication events that occur in hepatocytes. JFH-1-transfected Huh7.5.1 cells release up to 105 focus-forming units per mL. This number is 50-fold higher as compared to Huh7 cells. The difference appears to be due in part to higher permissiveness of Huh7.5.1 cells. Several features may explain this high permissiveness: i) Huh7.5.1 cells have a defect in the RIG-I pathway, making them less responsive to intracellular viral dsRNA; ii) they may express more numerous or “adequate” cell surface receptors mediating more efficacious HCV attachment and entry; or iii) virus assembly and budding is more effective in these cells.
Our study, which demonstrates that IFN fails to trigger PKR activation in non-infected cells and that both CypI and DAAs block PKR activation in HCV-infected cells, strongly supports the concept that suppression of viral replication results in the prohibition of the activation of IFN-mediated PKR during an established HCV infection. Our data also suggest that CypI in this experimental in vitro HCV infectivity design do not exert an action distinct from that of DAAs, at least in terms of IFN-induced PKR activation.
In conclusion, our study indicates that CypI do not have a unique effect on PKR activation, but rather the suppression of HCV replication by any anti-HCV agents, abrogates PKR activation. It also suggests that HCV exploits its dsRNA intermediates to trigger PKR activation that leads to suppression of translation of potentially antiviral ISGs that would otherwise endanger its survival. This study may also suggest that HCV hijacks PKR from the cellular antiviral machinery to weaken both the IFN-mediated antiviral response and IFN induction itself. Nevertheless, further work is required to determine whether PKR activation by HCV plays both a proviral and antiviral role during the IFN response depending on the phase of the infection (i.e., early versus established infection). It is important to re-emphasize that in this experimental infection cell culture system, IFN inhibits HCV replication despite PKR activation. We thus do not know at this stage whether the IFN-induced and HCV-dependent PKR activation is beneficial or detrimental to the virus. It would be extremely interesting to examine the degree of hepatic PKR expression and activation in HCV-infected patients prior to, during and after either an IFN treatment or an IFN-free regimen composed of a combination of DAAs and CypI.
ABBREVIATIONS
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
We thank Drs. Wakita and Chisari for Huh-7.5.1 and GT2a subgenomic JFH-1 replicon cells, Dr. Rice for the 9E10 anti-NS5A antibody, Drs. Bukh and Rice for the GT3a and 4a subgenomic JFH-1 replicon cells, Drs. Wakita and Pietschmann for the genomic Luc-JFH-1 plasmid, Novartis for alisporivir and Drs. Wilkinson and Gregory for cyclosporine A, sanglifehrins A and B. This work was supported by the U.S. Public Health Service grant no. AI087746 from the National Institute of Allergy and Infectious Diseases (NIAID). This is publication no. 25094 from the Department of Immunology & Microbial Science, The Scripps Research Institute, La Jolla, CA.