- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
HTS-Compatible Patient-Derived Cell-Based Assay to Identify Small Molecule Modulators of Aberrant Splicing in Myotonic Dystrophy Type 1
Debra A. O’Leary*, +, Leonardo Vargas, Orzala Sharif, Michael E. Garcia, Yury J. Sigal, Siu-Kei Chow, Christian Schmedt, Jeremy S. Caldwell#, Achim Brinker, Ingo H. Engels*
Abstract
Myotonic dystrophy type 1 (DM1) is a genetic disorder characterized by muscle wasting, myotonia, cataracts, cardiac arrhythmia, hyperinsulinism and intellectual deficits, and is caused by expansion of a CTG repeat in the 3’UTR of the Dystrophia Myotonica-Protein Kinase (DMPK) gene. The DMPK transcripts containing expanded CUG repeats accumulate in nuclear foci and ultimately cause mis-splicing of secondary genes through the dysregulation of RNA-binding proteins including Muscleblind 1 (MBNL1) and CUG binding protein 1 (CUGBP1). Correction of mis-splicing of genes such as the Skeletal muscle-specific chloride channel 1 (CLCN1), Cardiac troponin T (TNNT2), Insulin receptor (INSR) and Sarcoplasmic/endoplasmic reticulum Ca2+ATPase 1 (SERCA1) may alleviate some of the symptoms of DM1; hence identification of small molecule modulators is an important step towards a therapy for DM1 patients. Here we describe the generation of immortalized myoblast cell lines derived from healthy (DMPK CTG5) and DM1 patient (DMPK CTG1000) fibroblasts by constitutive overexpression of human telomerase reverse transcriptase (hTERT) and inducible overexpression of the Myoblast determination factor (MYOD). MBNL1-containing nuclear foci, mis-splicing events and defective myotube differentiation defects characteristic of DM1 were observed in these cells. A CLCN1 luciferase minigene construct (CLCN1-luc) was stably introduced to monitor intron 2 retention in the DM1 cellular context (a reported splicing defect in DM1). The assay was validated by performing a high-throughput screen (HTS) of ~13,000 low molecular weight compounds against the CLCN1-luc DM1 myoblast cell line, providing an ideal system for conducting HTS to better understand and treat DM1.
Article Information
Identifiers and Pagination:
Year: 2010Volume: 4
First Page: 9
Last Page: 18
Publisher Id: CCGTM-4-9
DOI: 10.2174/1875397301004010009
Article History:
Received Date: 9/9/2009Revision Received Date: 16/12/2009
Acceptance Date: 17/12/2009
Electronic publication date: 19/3/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to these authors at the Genomics Institute of the Novartis Research Foundation, 10675 John Jay Hopkins Drive, San Diego, CA 92121, USA; Tel: +1 (858) 812-1529; Fax: +1 (858) 812-1597; E-mails: oleary.debra@gmail.com; iengels@gnf.org+ Present address: Dart Neuroscience, San Diego, California, USA.
# Present address: Merck Research Laboratories, Department of Automated Biotechnology, North Wales, Pennsylvania, United States of America.
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 9-9-2009 |
Original Manuscript | HTS-Compatible Patient-Derived Cell-Based Assay to Identify Small Molecule Modulators of Aberrant Splicing in Myotonic Dystrophy Type 1 | |
INTRODUCTION
Myotonic dystrophy (DM) is the most common form of muscular dystrophy observed in adults, and symptoms include delayed relaxation of skeletal muscles following voluntary contraction (myotonia), muscle wasting, cataracts, cardiac arrhythmia, insulin resistance, and cognitive impairment [1Machuca-Tzili L, Brook D, Hilton-Jones D. Clinical and molecular aspects of the myotonic dystrophies: a review Muscle Nerve 2005; 32: 1-18.]. 98% of DM cases result from autosomal dominant inheritance of a CTG trinucleotide repeat expansion in the 3´UTR of the Dystrophia Myotonica-Protein Kinase (DMPK) gene [2Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene Science 1992; 255: 1253-5., 3Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member Cell 1992; 69: 385.] and are classified as DM type 1 (DM1). There is a correlation between the number of DMPK 3´UTR CTG repeats, age of onset and disease severity, with mildly affected patients having as few as 50 repeats and more acute cases ≥1000 repeats [3Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member Cell 1992; 69: 385.-5Gharehbaghi-Schnell EB, Finsterer J, Korschineck I, Mamoli B, Binder BR. Genotype-phenotype correlation in myotonic dystrophy Clin Genet 1998; 53: 20-6.]. Several mouse models expressing expanded CUG repeats (CUGexp) in the 3´UTR of unrelated mRNAs develop myotonia and myopathy [6Mankodi A, Logigian E, Callahan L, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat Science 2000; 289: 1769-73.], and DMPK knockout mice show only a subset of symptoms of DM1 [7Reddy S, Smith DB, Rich MM, et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy Nat Genet 1996; 13: 325-5.]. Together this indicates that it is not loss of DMPK function that causes the phenotypes of DM1, but instead an RNA gain of function.
DMPK CUGexp transcripts form hairpin structures that are retained in nuclear foci in DM1 cells [8Napierala M, Krzyzosiak WJ. CUG repeats present in myotonin kinase RNA form metastable "slippery" hairpins J Biol Chem 1997; 272: 31079-85.-10Furling D, Lam le T, Agbulut O, Butler-Browne GS, Morris GE. Changes in myotonic dystrophy protein kinase levels and muscle development in congenital myotonic dystrophy Am J Pathol 2003; 162: 1001-9.]. The majority of symptoms associated with DM1 are thought to result from the aberrant splicing of downstream target genes [1Machuca-Tzili L, Brook D, Hilton-Jones D. Clinical and molecular aspects of the myotonic dystrophies: a review Muscle Nerve 2005; 32: 1-18.]. Best studied are the dysregulation of the splicing factors MBNL1 and CUGBP1, which either alone or in concert are sufficient to cause many of the splicing defects associated with DM1 [11Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy Hum Mol Genet 2006; 15: 2087-97., 12Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy Science 1998; 280: 737-41.]. MBNL1 is normally involved in promoting muscle differentiation via regulation of postnatal changes in skeletal muscle splicing patterns [11Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy Hum Mol Genet 2006; 15: 2087-97., 13Pascual M, Vicente M, Monferrer L, Artero R. The muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing Differentiation 2006; 74: 65-80.]. In DM1, MBNL1 binds to and is sequestered into nuclear foci by the mutant DMPK transcripts [14Holt I, Jacquemin V, Fardaei M, et al. Muscleblind-like proteins: similarities and differences in normal and myotonic dystrophy muscle Am J Pathol 2009; 174: 216-7., 15Miller JW, Urbinati CR, Teng-Umnuay P, et al. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy EMBO J 2000; 19: 4439-8.]. The effect of MBNL1 loss of function is reflected in Mbnl1 knockout mice that exhibit Skeletal muscle-specific chloride channel 1 (Clcn1), Cardiac troponin T (Tnnt2) and Skeletal troponin T (Tnnt3) splicing abnormalities characteristic of DM1 [16Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy Science 2003; 302: 1978-80.]. The importance of MBNL1 function for maintenance of adult splicing patterns is highlighted by the fact that viral overexpression of Mbnl1 in skeletal muscle of a CUGexp mouse model promotes adult splicing patterns for Clcn1, Tnnt3 and Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 1(Serca1) [17Kanadia RN, Shin J, Yuan Y, et al. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy Proc Natl Acad Sci USA 2006; 103: 11748-53.]. Conversely, the consequences of increased CUGBP1 stability in DM1 are demonstrated in mice transgenic for human CUGBP1 in heart and skeletal muscle that show muscle degeneration and a shift towards fetal splicing patterns of several target genes [18Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy Hum Mol Genet 2005; 14: 1539-47.]. Taken together this data suggests that identification of regulators of aberrant splicing of MBNL1/CUGBP1 target genes may aid in development of therapies targeting the underlying cause of DM1.
Patients suffering from DM1 have traditionally been limited to symptomatic treatments including anticonvulsants and pain killers for myotonia, ventilators and pacemakers to improve respiratory and cardiac function, and physical exercise to fight muscle wasting. However, since the molecular mechanisms of DM1 etiology have been unraveled, the goal of researchers has shifted to the identification of more specific therapies, for which there is an unmet medical need. Progress is being made with antisense oligonucleotides and morpholinos targeting splice sites of the MBNL1/CUGBP1 target genes such as CLCN1 [19Wheeler TM, Lueck JD, Swanson MS, Dirksen RT, Thornton CA. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy J Clin Invest 2007; 117: 3952-7.] or the CUGexp transcript [20Mulders SA, van den Broek WJ, Wheeler TM, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy Proc Natl Acad Sci USA 2009; 106: 13915-20., 21Wheeler TM, Sobczak K, Lueck JD, et al. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA Science 2009; 325: 336-9.] in mouse models of DM1, but the exact mechanisms involved are still being elucidated, and delivery issues associated with intramuscular or intravenous injection of such therapies are yet to be resolved [22Cooper TA. Molecular biology. Neutralizing toxic RNA Science 2009; 325: 272-3.]. Small molecule compounds offer the advantage of oral formulation, and high-throughput screening (HTS) can be automated to assay millions of chemical entities in a short amount of time, even for somewhat complex cellular systems.
The key to the development of successful drug candidates from HTS hits depends largely upon the choice of the most relevant assay system for the given target [23Bender A, Bojanic D, Davies JW, et al. Which aspects of HTS are empirically correlated with downstream success? Curr Opin Drug Discov Devel 2008; 11: 327-7.]. Biochemical assays have the advantage of testing for direct interaction of chemicals with their target, however only cell-based assays allow for interrogation of pathways when the precise molecular target is unknown [24Moore K, Rees S. Cell-based versus isolated target screening: how lucky do you feel? J Biomol Screen 2001; 6: 69-74.]. Additional advantages of cell-based assays are the ability to monitor off-target effects, cell permeability and toxicity of compounds in the same system as was used for the primary screen. Previous assays designed to identify inhibitors of specific splice enhancers [25Soret J, Bakkour N, Maire S, et al. Selective modification of alternative splicing by indole derivatives that target serine-arginine-rich protein splicing factors Proc Natl Acad Sci USA 2005; 102: 8764-9.] or ligands for CUGexp RNA [26Pushechnikov A, Lee MM, Childs-Disney JL, et al. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3 J Am Chem Soc 2009; 131: 9767-79.] have been biochemical in nature and thus required secondary cell-based assays to test such factors. Given that aberrant splicing events in DM1 are both tissue- and developmental stage-specific in nature [11Lin X, Miller JW, Mankodi A, et al. Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy Hum Mol Genet 2006; 15: 2087-97., 27Kuyumcu-Martinez NM, Cooper TA. Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy Prog Mol Subcell Biol 2006; 44: 133-59.], and that skeletal muscle is the predominantly affected tissue in DM1 patients, one of the best cell types in which to develop an assay for monitoring aberrant splicing is patient-derived myoblasts.
To that end, we have generated immortalized fibroblast and myoblast cell lines derived from healthy (DMPK CTG5) and DM1 (DMPK CTG1000) individuals, as a tool for developing HTS-compatible assays in the DM1 cellular context. We stably introduced a CLCN1 luciferase minigene construct (CLCN1-luc) into these cells as a means of monitoring CLCN1 intron 2 retention. Aberrant inclusion of intron 2 and exon 7a in CLCN1 transcripts is thought to be the leading cause of myotonia in DM1 patients and mouse models [28Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy Mol Cell 2002; 10: 35-44., 29Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing Mol Cell 2002; 10: 45-53.], given that mutations in CLCN1 alone are associated with dominant and recessive myotonia [30Koch MC, Ricker K, Otto M, et al. Evidence for genetic homogeneity in autosomal recessive generalised myotonia (Becker) J Med Genet 1993; 30: 914-7., 31George AL Jr, Crackower MA, Abdalla JA, Hudson AJ, Ebers GC. Molecular basis of Thomsen's disease (autosomal dominant myotonia congenita) Nat Genet 1993; 3: 305-10.], and that transgenic mouse models with CUGexp in transcripts other than DMPK show similar patterns of CLCN1 mis-splicing and myotonia [28Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy Mol Cell 2002; 10: 35-44.]. We have miniaturized the CLCN1-luc assay into 1536-well format and conducted a HTS of ~13,000 small molecule compounds in triplicate, validating the DM1 patient-derived cell line and CLCN1-luc minigene reporter for use in larger screens to identify modulators of aberrant splicing and potential therapeutics for the treatment of DM1.
MATERIALS AND METHODOLOGY
Generation of Inducible MYOD Construct
The coding region of human MYOD was amplified from the MGC:8630 IMAGE:2961494 clone in pOTB7 backbone using PCR primers containing SrfI restriction sites (forward:5'CGCCAGCCCGGGCCCATGGAGCTACTGTCGCCAC3' and reverse:5' CGGCCAGCCCGGGCTCAGAGCACC3') and Phusion™ high fidelity DNA polymerase (NEB) as per manufacturer’s instructions. Following overnight digestion with SrfI restriction endonuclease (NEB) this PCR product was cloned into the SrfI sites of a pTRE-rtTA3Zeo-SrfccdBSrf vector which was modified from the pTREautoR3 vector [32Markusic D, Oude-Elferink R, Das AT, Berkhout B, Seppen J. Comparison of single regulated lentiviral vectors with rtTA expression driven by an autoregulatory loop or a constitutive promoter Nucleic Acids Res 2005; 33: e63.] (kindly provided by Dr. David Markusic) as follows. A Zeocin resistance gene cassette had flanking foot-and-mouth disease virus (FMDV) 2a peptide sequences added on either side (to allow for generation of a multigene vector) and was cloned in-frame with the tetracycline responsive transactivator ( rtTA) gene. The 5´ 2a sequence was designed to end with a SrfI restriction site. The counter-selectable suicide gene ccdB was cloned into the SrfI site, generating pTRE-rtTA3Zeo-SrfccdBSrf. The MYOD PCR product was cloned into the SrfI sites of pTRE-rtTA3Zeo-SrfccdBSrf replacing the ccdB gene and generating the MYOD/pTREautoR3 vector, which has the polycistronic open reading frame rtTA-2a-Zeo-2a-MYOD under control of the tetracycline responsive promoter (TRE). Correct orientation and reading frame were confirmed by sequencing.
Generation of CLCN1-luc Construct
The genomic segment spanning exon 2 to exon 3 of human CLCN1 (3630-4670 bp of GI:89161213) was amplified by PCR using NEB Phusion polymerase and 200 ng male genomic DNA as template (Promega), generating a 1 kb product with NcoI restriction sites at both ends (forward: 5´CTCTGCCATGGTTTATGGCCATCACAAAGAAC3´ and reverse: 5´GGTTAACCATGGCCTGAAGGCTTTTGGCA CTGAC3´). This fragment was cloned into the NcoI site immediately upstream of luciferase in the pGL3 promoter vector (Promega), and the ATG start codon of luciferase was mutated to TTG via single primer mutagenesis [33Shenoy AR, Visweswariah SS. Site-directed mutagenesis using a single mutagenic oligonucleotide and DpnI digestion of template DNA Anal Biochem 2003; 319: 335-6.] using primer sequence 5´AGCCTTCAGGCCTTGGAAGACGCC3´. The CLCN1-luc construct was generated by sub-cloning the CLCN1-luc cassette into the pLenti6/V5-D-TOPO vector (Invitrogen) as per manufacturer’s instructions. Correct orientation and reading frame were confirmed by sequencing.
Virus Production
The pBabe-puro vector expressing the catalytic subunit of human Telomerase reverse transcriptase (hTERT) was a kind gift from Dr. Robert Weinberg [34Counter CM, Hahn WC, Wei W, et al. Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization Proc Natl Acad Sci USA 1998; 95: 14723-8.]. Retrovirus expressing hTERT was produced by co-transfection with the packaging vector pCL-10A1. 10 µg of each vector was transfected into 5 x 106 293T cells in a 10 cm tissue culture dish (Greiner) using FuGENE™ 6 (Roche) as per manufacturer’s instructions. 24 h post-transfection cells were re-fed with growth media consisting of DMEM (Gibco®) supplemented with 10% fetal bovine serum (Hyclone) and 1X antibiotic-antimycotic (Gibco®). Viral supernatant was harvested and filtered through a 0.45 µm filter (Millipore) 48 h post-transfection.
Lentivirus expressing MYOD or CLCN1-luc was produced by co-transfection of 10 µg MyoD/pTREautoR3 or CLCN1-luc/pLenti6/V5-D-TOPO DNA with RRE, REV and VSV-g packaging vectors (6.5 µg, 2.5 µg and 3.5 µg, respectively) into 293Tcells, and viral supernatant was harvested as described above.
Generation of Immortalized Myoblast and CLCN1-luc Cell Lines
Primary fibroblast cell lines GM00321 (DMPK CTG5) and GM04033 (DMPK CTG1000) were purchased from Coriell Institute for Medical Research and maintained in MEM alpha growth media (Gibco®) supplemented with 15% fetal bovine serum (FBS) (Hyclone) and 1X antibiotic-antimycotic (Gibco®). Cells were immortalized by viral infection with supernatant expressing hTERT. Briefly, 1 x 106 cells were seeded in a T75 tissue culture flask (Greiner) and the next day treated with 2 ml of viral supernatant for 6 h prior to supplementation with 10 ml growth media. 48 h post-infection media was replaced and supplemented with 1 µg/ml puromycin (Hyclone). Selection was maintained for 5 days prior to cell expansion. Immortalized cell lines were subsequently infected with MYOD supernatant and selected for 14 days in growth media supplemented with 200 µg/ml zeocin (Invitrogen) and 5 µg/ml doxycycline (Sigma). The CLCN1-luc reporter gene was introduced by infection of hTERT positive, inducible MYOD positive cells with CLCN1-luc supernatant and selection for 5 days with 1 µg/ml blasticidin (Invitrogen).
Following recovery from selection, immortalized wildtype and DM1 fibroblasts were differentiated into myoblasts upon treatment with 5 µg/ml doxycycline (Sigma) for at least 24 h, or into myotubes after 3-7 days growth in differentiation media (DMEM supplemented with 2% horse serum (Hyclone) and 1X antibiotic-antimycotic (Gibco®)) containing 5 µg/ml doxycycline. All cells were maintained in 37oC incubators with 5% CO2.
RT-PCR Analysis
Total RNA was extracted from ~200,000 cultured cells grown in Greiner 6-well tissue culture treated plates using Qiagen RNeasy mini kit columns and quantified using a NanoDrop 1000 spectrophotometer (Thermo Scientific). 200 ng RNA was used as template for cDNA synthesis with random primers and SuperScript® II reverse transcriptase (Invitrogen), as per manufacturer’s instructions. 1 µl of the resulting cDNA was used as template for each RT-PCR with Platinum® Taq DNA polymerase (Invitrogen). The following primers were used: hTERT-pBabe3´UTR forward - 5'GAGGTGCAGAGCGACTACTC3' and reverse - 5'CACCC TAACTGACACACATTCC3'; TNNT2 forward - 5'ATAGAA GAGGTGGTGGAAGAGTAC3' and reverse - 5'GTCTCAGC CTCTGCTTCAGCATCC3'; INSR forward - 5'CCAAAGA CAGACTCTCAGAT3' and reverse - 5'AACATCGCCAAG GGACCTGC3'; SERCA1 forward - 5'GATGATCTTC AAGCTCCGGGC3' and reverse - 5'CAGCTCTGCCTGA AGATGTG3'; CLCN1-luc forward - 5´CTCTGCCATGGTTT ATGGCCATCACAAAGAAC3´ and reverse - 5´AGGGCGT ATCTCTTCATAGCCTTA3´; GAPDH forward - 5'TGCACC ACCAACTGCTTA3' and reverse - 5'GGATGCAGGG ATGATGTTC3'. RT-PCR was performed for 35 cycles using standard PCR conditions, annealing temperature of 55oC and extension time of 30-60 seconds. PCR products were visualized by the addition of SYBR® Gold (Invitrogen) to samples prior to electrophoresis at 100 V using 2-3% agarose (Sigma) dissolved in Tris-acetate-EDTA buffer (Sigma) and density of bands quantified using Alpha Innotech’s AlphaEase© FC software, version 3.2.1 (values expressed as average ± SEM).
Western Blotting
Total and cytoplasmic/nuclear cell extracts were prepared by lysing cells directly in 15 cm tissue culture dishes (Greiner) as per manufacturer’s instructions (Active Motif). Samples were quantified by addition of Coomassie Protein Assay Reagent (Pierce) and readout with a Molecular Devices SpectraMax Plus384 plate reader at 595 nm. Protein samples (20-40 µg per lane) were subjected to SDS-PAGE using NuPAGE® Novex Bis-Tris 4-12% gradient precast gels (Invitrogen), and subsequently transferred onto 0.45 µm nitrocellulose membrane (Invitrogen), as per manufacturer’s instructions. After incubation at room temperature with Blocker Casein in TBS (Pierce) for 1-2 h, membranes were incubated with antibody at 4oC overnight (with shaking). Antibodies used were specific to MYOD (1:500 sc-760 – Santa Cruz), CLCN1 (1:1000 CLC11-A – Alpha Diagnostic), CUG-BP1 (1:1000 05-621 – Upstate), MBNL1 (1:100 ab49474 - abcam), LAMIN A/C (1:500 sc-20681 Santa Cruz), and β-ACTIN (1:5000 A5316 – Sigma), and were diluted in blocking buffer supplemented with 0.1% Tween-20 (Sigma). Following washing in Tris-buffered saline with Tween-20 (TBST) (Sigma), membranes were incubated with secondary antibodies conjugated with IRDye™800 (Rockland) or Alexa Fluor®680 dye (Invitrogen) for 1 h at room temperature (1:20,000 dilution in blocking buffer supplemented with 0.1% Tween-20), washed again with TBST and visualized (and protein expression quantified) using a LI-COR Odyssey® infrared imaging system. Quantified protein levels are expressed as average ± SEM.
In Situ Hybridization and Immunostaining
Method adapted from Mankodi et al. 2001 [35Mankodi A, Urbinati CR, Yuan QP, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2 Hum Mol Genet 2001; 10: 2165-70.]. Cells were grown in 39 mm cell culture dishes with 20 mm coverglass insets (In vitro Scientific), fixed with 4% paraformaldehyde (PFA) (Electron Microscopy Sciences) at room temperature for 20 minutes, washed in phosphate-buffered saline (PBS) (Sigma), and permeabilized by treatment with ice-cold 2% acetone (Fisher Scientific) for 5 minutes. After PBS washing cells were pre-hybridized for 1 h at 45oC in a wet chamber with distilled water using a buffer of 30% formamide (Sigma) and 2X saline-sodium citrate buffer (SSC) (Sigma). Hybridization was then performed in a light-proof wet chamber for 2 h at 45oC using the same buffer supplemented with 0.02% bovine serum albumin (BSA) (Sigma), 66 µg/ ml yeast tRNA (Sigma), 2 mM sodium (meta)vanadate (Sigma) and 1 ng/µl RNase-free HPLC-purified (CAG)7 2' O-methyl RNA probe with a phosphorothioate backbone and 5' Alexa Fluor® 594 label (Integrated DNA Technologies). Following hybridization cells were washed 3 x 10 minutes at 45oC in a solution of 30% formamide and 2X SSC, then 3 x 10 minutes at room temperature (with shaking) in a 1X SSC solution. MBNL1 immunostaining was then performed on these samples by blocking with a solution of 1% FBS and 1% BSA in PBS for 1 h at room temperature and incubation with 2 µg/ ml primary antibody (in PBS) at 4oC overnight (M3320 – Sigma). Antibody was washed off with 5 x 5 minute PBS washes and 1:500 Alexa Fluor®488 donkey anti-mouse secondary antibody (Invitrogen) was added for 2 h at room temperature. Following 5 x 5 minute PBS washes, cells were incubated with 7 µg/ ml Hoechst 33342 (Invitrogen) in PBS for at least 1 h at room temperature prior to imaging with a 60x/1.20W Olympus UPlanSApo objective on a Perkin Elmer UltraVIEW VoX Confocal imaging system.
Immunostaining for Desmin (1:50 18-0016 - Invitrogen) and Myosin heavy chain (MHC) (1:250 18-0105 Invitrogen) was performed as for MBNL1 after fixation of cells in 24 well plates (Greiner) in 2% PFA and permeabilization with 0.25% Triton® X-100 in PBS for 5 minutes. In order to visualize cell morphology 20 µg/ml malachite green (Sigma) was added in parallel to Hoechst. Cells were imaged with a 20x/0.40 Olympus LCPlanFI objective on a Perkin Elmer UltraVIEW VoX Confocal imaging system.
CLCN1-luc Reporter Gene Assay and HTS
Stable CLCN1-luc DM1 or WT cells were plated at a density of 8000 cells/well in 50 µl for 384-well format or 2000 cells/well in 6 µl for 1536-well format (in the presence of doxycycline) in tissue culture-treated Greiner custom white solid bottom plates using GNF Systems on-line screening equipment, incubated at 37oC with 5% CO2 for 24 h prior to 500 nl (384-well) or 50 nl (1536-well) compound addition with a PinTool (GNF Systems) to give 1 % or 0.83% DMSO, respectively. Luciferase activity was then measured 24 h post-compound addition by the addition of 30 µl (384-well) or 3 µl (1536-well) Bright-Glo (Promega) and a 60 second luminescence read with a ViewluxTM CCD Imager (Perkin Elmer). For the HTS of ~13,000 compounds in triplicate, CLCN1-luc DM1 cells were treated as described above for 1536-well format such that the final compound concentration was 8.3 µM, and the AKT V inhibitor Triciribine (Calbiochem) was included on each assay plate as a positive control.
RESULTS AND DISCUSSION
Generation of Immortalized Myoblasts with Differentiation Defects Characteristic of DM1
Given the complex nature of DM1 etiology in humans, and the fact that single gene transgenic and knockout mouse models of DM1 fail to recapitulate all symptoms of the human disease [6Mankodi A, Logigian E, Callahan L, et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat Science 2000; 289: 1769-73., 16Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy Science 2003; 302: 1978-80., 18Ho TH, Bundman D, Armstrong DL, Cooper TA. Transgenic mice expressing CUG-BP1 reproduce splicing mis-regulation observed in myotonic dystrophy Hum Mol Genet 2005; 14: 1539-47., 36Jansen G, Groenen PJ, Bachner D, et al. Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice Nat Genet 1996; 13: 316-24.-42Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis J Biol Chem 2004; 279: 13129-39.], we chose to generate immortalized DM1 patient-derived cell lines in which to develop a HTS-amenable assay. This would also allow for direct inference of assay results to the human disease condition, since signaling pathways are not always identical between mouse and human. Wildtype (WT) DMPK CTG5 and DM1 patient (DMPK CTG1000) fibroblast cells were immortalized following introduction of constitutively overexpressed hTERT, which has previously been shown to allow human fibroblasts to proliferate for at least 36 population doublings [43Iujvidin S, Fuchs O, Nudel U, Yaffe D. SV40 immortalizes myogenic cells: DNA synthesis and mitosis in differentiating myotubes Differentiation 1990; 43: 192-203.]. RT-PCR confirmed expression of hTERT in immortalized WT and DM1 cell lines carrying hTERT alone (hTERT FB) or hTERT and MYOD expression constructs (MYOD FB), and was maintained upon differentiation of these cells into myoblasts and myotubes with doxycycline treatment and serum starvation (Fig. 1A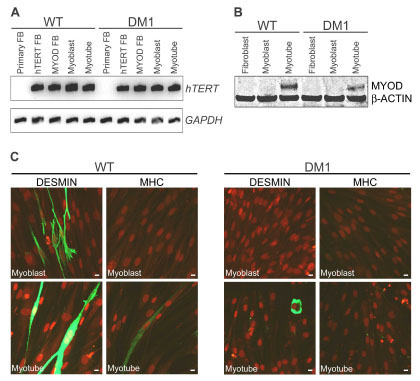 ). Use of fibroblasts as a starting point allowed monitoring of myogenic differentiation defects in DM1 cells upon inducible overexpression of MYOD, the sole transcription factor necessary to convert fibroblast cells into myoblasts [44Tapscott SJ, Davis RL, Thayer MJ, Cheng PF, Weintraub H, Lassar AB. MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts Science 1988; 242: 405-11.].
). Use of fibroblasts as a starting point allowed monitoring of myogenic differentiation defects in DM1 cells upon inducible overexpression of MYOD, the sole transcription factor necessary to convert fibroblast cells into myoblasts [44Tapscott SJ, Davis RL, Thayer MJ, Cheng PF, Weintraub H, Lassar AB. MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts Science 1988; 242: 405-11.].

As shown in Fig. (1B), MYOD protein expression was barely detectable by Western blot 24 h after treatment of immortalized fibroblasts with doxycycline (myoblasts), but was significantly increased upon differentiation of myoblasts into myotubes for 3-7 days. Upon quantification of Western blots across multiple experiments, DM1 myotubes were observed to express between 63±4% of WT MYOD protein levels (relative to β-actin housekeeping protein). A similar trend was observed by RT-PCR (data not shown). Decreased MYOD expression has previously been reported in cells overexpressing transcripts with a CUGexp [45Amack JD, Reagan SR, Mahadevan MS. Mutant DMPK 3'-UTR transcripts disrupt C2C12 myogenic differentiation by compromising MyoD J Cell Biol 2002; 159: 419-29.], and more recently in muscle biopsy samples of DM1 patients [46Zhao XP, Pu CQ, Duan F, Liu JX, Mao YL, Luo P. MyoD mRNA expression in skeletal muscle of patients with myotonic dystrophy Zhonghua Yi Xue Za Zhi 2009; 89: 466-8.]. As would be expected from reduced expression of MYOD, DM1 myoblasts showed limited capacity to differentiate down the myogenic lineage, as tested by immunostaining for the early myogenic marker Desmin [47Carter RL, McCarthy KP, Machin LG, Jameson CF, Philp ER, Pinkerton CR. Expression of desmin and myoglobin in rhabdomyosarcomas and in developing skeletal muscle Histopathology 1989; 15: 585-95.] and late myogenic differentiation marker MHC [48Andres V, Walsh K. Myogenin expression, cell cycle withdrawal, and phenotypic differentiation are temporally separable events that precede cell fusion upon myogenesis J Cell Biol 1996; 132: 657-6.], together with malachite green counterstaining for morphological changes in comparison to WT cells (Fig. 1C). In vitro differentiation of myoblasts into myotubes is a highly variable, fluid process affected by cell density and growth conditions at the time of serum starvation, so variable morphology and stages of differentiation are often observed within one culture. Overexpression of hTERT in these cell lines may have had some effect on the differentiation potential of WT and DM1 cells, as hTERT is usually down regulated during myoblast differentiation [49Ma H, Urquidi V, Wong J, Kleeman J, Goodison S. Telomerase reverse transcriptase promoter regulation during myogenic differentiation of human RD rhabdomyosarcoma cells Mol Cancer Res 2003; 1: 739-46.]. However, since a significant distinction in differentiation potential (lack of Desmin expression) was reproducibly observed between WT and DM1 myoblasts prior to serum starvation, the immortalized DM1 cell lines were further characterized for their suitability to study aberrant splicing events.
Immortalized Cell Lines Show MBNL1 Localization and Splicing Defects Characteristic of DM1
Immortalized WT and DM1 cell lines were analyzed by Western blot for expression of the splicing factors CUGBP1 and MBNL1 (Fig. 2A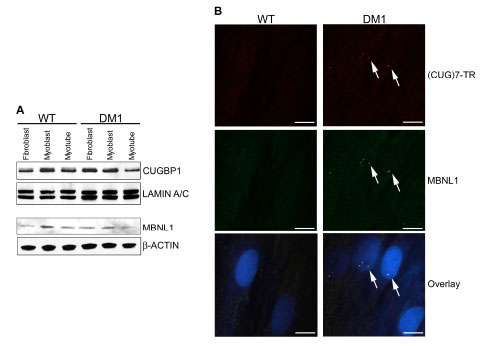 ). No significant difference in protein expression levels was observed between genotypes or throughout differentiation of the immortalized WT and DM1 fibroblasts into myoblasts and myotubes. Altered cellular distribution, increased expression levels and hyperphosphorylation of CUGBP1 protein have been reported in some DM1 patients and mouse models [50Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1 J Biol Chem 2001; 276: 7820-6., 51Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation Mol Cell 2007; 28: 68-78.], however decreased CUGBP1 mRNA levels were reported in DM1 muscle biopsies [52Watanabe T, Takagi A, Sasagawa N, Ishiura S, Nakase H. Altered expression of CUG binding protein 1 mRNA in myotonic dystrophy 1: possible RNA-RNA interaction Neurosci Res 2004; 49: 47-54.]. In addition, MBNL1 localization to nuclear foci containing CUGexp transcripts is a more definitive trait of DM1 cells [35Mankodi A, Urbinati CR, Yuan QP, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2 Hum Mol Genet 2001; 10: 2165-70., 53Cardani R, Mancinelli E, Rotondo G, Sansone V, Meola G. Muscleblind-like protein 1 nuclear sequestration is a molecular pathology marker of DM1 and DM2 Eur J Histochem 2006; 50: 177-82.], so cells were further analyzed for MBNL1 cellular distribution. MBNL1 protein was found to co-localize with CUGexp transcripts in nuclear foci in immortalized DM1 myoblasts (Fib. 2B), as well as DM1 fibroblasts and myotubes (data not shown). Immunostaining revealed that MBNL1 protein was evenly distributed throughout the nuclei of WT cells (data not shown), however as previously described [14Holt I, Jacquemin V, Fardaei M, et al. Muscleblind-like proteins: similarities and differences in normal and myotonic dystrophy muscle Am J Pathol 2009; 174: 216-7.], the acetone treatment required for in situ hybridization prevented visualization of this in the same experiment.
). No significant difference in protein expression levels was observed between genotypes or throughout differentiation of the immortalized WT and DM1 fibroblasts into myoblasts and myotubes. Altered cellular distribution, increased expression levels and hyperphosphorylation of CUGBP1 protein have been reported in some DM1 patients and mouse models [50Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1 J Biol Chem 2001; 276: 7820-6., 51Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation Mol Cell 2007; 28: 68-78.], however decreased CUGBP1 mRNA levels were reported in DM1 muscle biopsies [52Watanabe T, Takagi A, Sasagawa N, Ishiura S, Nakase H. Altered expression of CUG binding protein 1 mRNA in myotonic dystrophy 1: possible RNA-RNA interaction Neurosci Res 2004; 49: 47-54.]. In addition, MBNL1 localization to nuclear foci containing CUGexp transcripts is a more definitive trait of DM1 cells [35Mankodi A, Urbinati CR, Yuan QP, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2 Hum Mol Genet 2001; 10: 2165-70., 53Cardani R, Mancinelli E, Rotondo G, Sansone V, Meola G. Muscleblind-like protein 1 nuclear sequestration is a molecular pathology marker of DM1 and DM2 Eur J Histochem 2006; 50: 177-82.], so cells were further analyzed for MBNL1 cellular distribution. MBNL1 protein was found to co-localize with CUGexp transcripts in nuclear foci in immortalized DM1 myoblasts (Fib. 2B), as well as DM1 fibroblasts and myotubes (data not shown). Immunostaining revealed that MBNL1 protein was evenly distributed throughout the nuclei of WT cells (data not shown), however as previously described [14Holt I, Jacquemin V, Fardaei M, et al. Muscleblind-like proteins: similarities and differences in normal and myotonic dystrophy muscle Am J Pathol 2009; 174: 216-7.], the acetone treatment required for in situ hybridization prevented visualization of this in the same experiment.

The effect of MBNL1 mis-localization upon splicing of downstream target genes in immortalized DM1 cells was tested by RT-PCR using primers specific for three of the best characterized DM1 splicing defects in TNNT2, SERCA1 and INSR genes. These mis-splicing events are thought to contribute to the cardiac arrhythmia and insulin resistance observed in DM1 patients [1Machuca-Tzili L, Brook D, Hilton-Jones D. Clinical and molecular aspects of the myotonic dystrophies: a review Muscle Nerve 2005; 32: 1-18.]. The splicing patterns observed in the WT and DM1 cell lines were as expected (Fig. 3A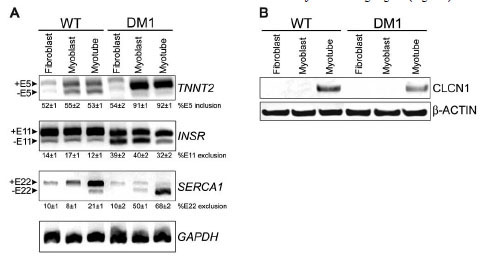 ): exon 5 was included in 91±1% and 92±1% of TNNT2 transcripts in DM1 myoblasts and myotubes, respectively (versus 55±2 and 53±1% in WT counterparts); exon 11 was excluded in 39±2% and 40±2% of INSR transcripts in DM1 fibroblasts and myoblasts, respectively (versus 14±1% and 17±1% in WT counterparts); exon 22 was excluded in 50±1% and 68±2% of SERCA1 transcripts in DM1 myoblasts and myotubes, respectively (versus 8±1% and 21±1% in WT counterparts). Mis-splicing and expression loss of CLCN1 protein in skeletal muscle is associated with myotonia in DM1 patients and mouse models [28Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy Mol Cell 2002; 10: 35-44., 29Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing Mol Cell 2002; 10: 45-53.]. Since CLCN1 mRNA expression in cultured myoblasts is ≤ 0.01% of that in mature muscle tissue [54Bardouille C, Vullhorst D, Jockusch H. Expression of chloride channel 1 mRNA in cultured myogenic cells: a marker of myotube maturation FEBS Lett 1996; 396: 177-80.], we tested for CLCN1 protein expression levels in our immortalized WT and DM1 cell lines by Western blot (Fig. 3B). CLCN1 protein expression could only be detected in differentiated myotubes, and the expression level in DM1 myotubes was 43±1% of WT levels (determined across multiple experiments, relative to β-actin loading control). This is consistent with previous findings in DM1 patient tissue and mouse models [28Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy Mol Cell 2002; 10: 35-44.]. Therefore the immortalized DM1 myoblast cell line was found to retain the aberrant splicing and expression loss of MBNL1/CUGBP1 target genes, and the myogenic differentiation defect associated with the disease. We proceeded to use this tool for development of a HTS-compatible assay to monitor mis-splicing in DM1.
): exon 5 was included in 91±1% and 92±1% of TNNT2 transcripts in DM1 myoblasts and myotubes, respectively (versus 55±2 and 53±1% in WT counterparts); exon 11 was excluded in 39±2% and 40±2% of INSR transcripts in DM1 fibroblasts and myoblasts, respectively (versus 14±1% and 17±1% in WT counterparts); exon 22 was excluded in 50±1% and 68±2% of SERCA1 transcripts in DM1 myoblasts and myotubes, respectively (versus 8±1% and 21±1% in WT counterparts). Mis-splicing and expression loss of CLCN1 protein in skeletal muscle is associated with myotonia in DM1 patients and mouse models [28Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy Mol Cell 2002; 10: 35-44., 29Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing Mol Cell 2002; 10: 45-53.]. Since CLCN1 mRNA expression in cultured myoblasts is ≤ 0.01% of that in mature muscle tissue [54Bardouille C, Vullhorst D, Jockusch H. Expression of chloride channel 1 mRNA in cultured myogenic cells: a marker of myotube maturation FEBS Lett 1996; 396: 177-80.], we tested for CLCN1 protein expression levels in our immortalized WT and DM1 cell lines by Western blot (Fig. 3B). CLCN1 protein expression could only be detected in differentiated myotubes, and the expression level in DM1 myotubes was 43±1% of WT levels (determined across multiple experiments, relative to β-actin loading control). This is consistent with previous findings in DM1 patient tissue and mouse models [28Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy Mol Cell 2002; 10: 35-44.]. Therefore the immortalized DM1 myoblast cell line was found to retain the aberrant splicing and expression loss of MBNL1/CUGBP1 target genes, and the myogenic differentiation defect associated with the disease. We proceeded to use this tool for development of a HTS-compatible assay to monitor mis-splicing in DM1.

CLCN1-luc Minigene Reporter Assay Validation
Myotonia is one of the most prevalent symptoms of DM1 [1Machuca-Tzili L, Brook D, Hilton-Jones D. Clinical and molecular aspects of the myotonic dystrophies: a review Muscle Nerve 2005; 32: 1-18., 55Logigian EL, Ciafaloni E, Quinn LC, et al. Severity, type, and distribution of myotonic discharges are different in type 1 and type 2 myotonic dystrophy Muscle Nerve 2007; 35: 479-85.], and aberrant inclusion of intron 2 and exon 7a of the skeletal muscle-specific chloride channel gene CLCN1 is associated with the myotonia observed in DM1 patients and mouse models [28Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy Mol Cell 2002; 10: 35-44., 29Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing Mol Cell 2002; 10: 45-53.]. Therefore we chose to generate a luciferase minigene reporter construct spanning the genomic region of exon 2 to exon 3 of human CLCN1, driven by the CMV promoter of the pLenti6/V5-D-TOPO vector (Invitrogen). As shown by the schematic in Fig. (4A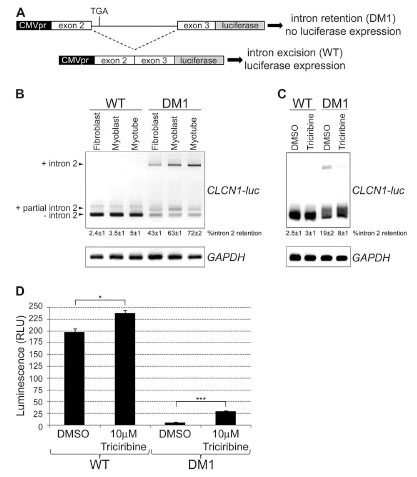 ), the correct reading frame of the minigene was obtained upon removal of intron 2 during splicing in WT cells, however intron 2 retention in DM1 patient cells would lead to use of a premature stop codon and lack of luciferase expression. RT-PCR analysis of WT and DM1 CLCN1-luc stable cell lines with primers spanning from CLCN1 exon 2 to the beginning of the luciferase coding region (Fig. 4B) demonstrated that the minigene reporter construct expressed as expected. Complete intron 2 retention was found in 43±1%, 63±1% and 72±2% of CLCN1-luc transcripts in DM1 fibroblasts, myoblasts and myotubes, respectively (versus 2.4±1%, 3.5±1% and 5±1% in WT counterparts), and partial intron 2 retention was seen at similar levels (14±2%) in CLCN1-luc transcripts of both WT and DM1 cells, in agreement with a previously identified cryptic splice site in CLCN1 intron 2 [29Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing Mol Cell 2002; 10: 45-53.]. Identity of all RT-PCR products was confirmed by sequencing.
), the correct reading frame of the minigene was obtained upon removal of intron 2 during splicing in WT cells, however intron 2 retention in DM1 patient cells would lead to use of a premature stop codon and lack of luciferase expression. RT-PCR analysis of WT and DM1 CLCN1-luc stable cell lines with primers spanning from CLCN1 exon 2 to the beginning of the luciferase coding region (Fig. 4B) demonstrated that the minigene reporter construct expressed as expected. Complete intron 2 retention was found in 43±1%, 63±1% and 72±2% of CLCN1-luc transcripts in DM1 fibroblasts, myoblasts and myotubes, respectively (versus 2.4±1%, 3.5±1% and 5±1% in WT counterparts), and partial intron 2 retention was seen at similar levels (14±2%) in CLCN1-luc transcripts of both WT and DM1 cells, in agreement with a previously identified cryptic splice site in CLCN1 intron 2 [29Charlet BN, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing Mol Cell 2002; 10: 45-53.]. Identity of all RT-PCR products was confirmed by sequencing.

Since aberrant splicing patterns of MBNL1/CUGBP1 target genes were observed in both immortalized DM1 myoblasts and myotubes, further HTS assay development was limited to myoblasts, as this allowed for easier propagation of cells, shorter incubation times prior to luciferase readout, and was not complicated by the heterogeneity of myotube differentiation stage between wells and across experiments. A panel of protein kinase B (PKB or AKT) and protein kinase C (PKC) inhibitors were screened in 384-well format for their ability to serve as a positive control by enhancing expression of the CLCN1-luc construct in DM1 myoblasts (data not shown), based on reports that CUGBP1 activation and phosphorylation is mediated by members of these kinase families [51Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation Mol Cell 2007; 28: 68-78., 56Salisbury E, Sakai K, Schoser B, et al. Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1 Exp Cell Res 2008; 314: 2266-78.]. From these experiments only one compound, an AKT V inhibitor Triciribine (Calbiochem), enhanced luciferase expression in DM1 CLCN1-luc myoblasts by 5.3±0.4 fold 24 h after compound addition (p = 1.1 x 10-5 in a two-tailed t-test) (Fig. 4D), and RT-PCR analysis confirmed that this was due to partial restoration of CLCN1-luc splicing in treated DM1 cells (Fig. 4C). Treatment of cells with DMSO alone enhanced CLCN1-luc splicing in DM1 myoblasts, and was presumed to act by making RNA more accessible to general splicing factors. The splicing of CLCN1-luc was further improved following treatment of DM1 CLCN1-luc myoblasts with Triciribine; only 8±1% of transcripts contained intron 2 following Triciribine treatment (versus 19±2% in DMSO treated cells), and 59±2% of transcripts lacked intron 2 completely. Triciribine only gave a 1.2±0.2 fold increase in luciferase signal in CLCN1-luc WT myoblasts compared to DMSO treatment (p = 2.4 x 10-3), but this was not surprising given that these cells already spliced the reporter construct correctly. Given its reproducible effect on the CLCN1-luc construct in DM1 myoblasts, Triciribine was used as a positive control for miniaturization of the CLCN1-luc DM1 myoblast reporter gene assay into 1536-well format and assay validation for HTS.
Assay Miniaturization and HTS Validation of CLCN1-luc DM1 Myoblasts
The CLCN1-luc assay in DM1 myoblasts was miniaturized from a 50 µl cell plating volume in 384-well format to a 6 µl volume in 1536-well format. The health of the CLCN1-luc DM1 myoblasts was maintained with a final DMSO concentration up to 1% in both 384-well and 1536-well formats (as determined by alamarBlue® (BioSource™), data not shown), and 0.83% DMSO (50 nl into 6 µl) was used for the HTS validation study. As shown in Fig. (5A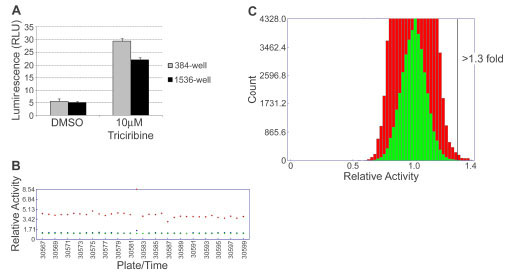 ), miniaturization of the assay lead to a slight decrease in assay window; 5.3 fold increase in signal upon Triciribine treatment in 384-well format and 4.3 fold increase in 1536-well format. For ease of comparison with test compounds and to prevent cellular toxicity, a final concentration of 8.3 µM Triciribine was used as a positive control on all 1536-well assay plates during the HTS validation, which was the same concentration as the compounds screened.
), miniaturization of the assay lead to a slight decrease in assay window; 5.3 fold increase in signal upon Triciribine treatment in 384-well format and 4.3 fold increase in 1536-well format. For ease of comparison with test compounds and to prevent cellular toxicity, a final concentration of 8.3 µM Triciribine was used as a positive control on all 1536-well assay plates during the HTS validation, which was the same concentration as the compounds screened.

The robustness of the CLCN1-luc assay was tested by performing a validation HTS of ~13,000 small molecule compounds in triplicate in 1536-well format. Fig. (5B) depicts the trace of plate median relative activity for test compound wells, as well as DMSO and Triciribine control wells, and shows maintenance of an assay window of 4-fold throughout the screen, with a Z´ factor = 0.5 and CV = 9%. Thus the CLCN1-luc assay was deemed amenable to HTS. By plotting a histogram of relative activity of test compounds (Fig. 5C) a normal distribution with mean = 1.0 was observed and a hit rate of 0.26% was observed using a threshold of greater than 1.3 fold increase in CLCN1-luc signal above that of the DMSO negative control. The reproducibility of hits was 29.8%, and may be improved in future studies by raising the threshold for hit determination.
CONCLUSION
We have generated an immortalized DM1 (DMPK CTG1000) fibroblast cell line that is easily induced to differentiate into myoblasts, and retains DM1 phenotypic characteristics including defective differentiation into myotubes, MBNL1 co-localization with CUGexp transcripts in nuclear foci, and mis-splicing of MBNL1/CUGBP1 target genes. In addition, we have developed the CLCN1-luc luciferase-based minigene reporter assay that monitors aberrant splicing of CLCN1 and shows the expected increase in intron 2 retention in immortalized DM1 myoblasts in comparison to WT cells. The CLCN1-luc assay in DM1 (DMPK CTG1000) myoblasts was successfully miniaturized into 1536-well format and shown to be robust enough for HTS, and the AKT inhibitor Triciribine was identified as a useful tool compound that partially restores CLCN1 splicing in DM1 cells. The CLCN1-luc construct was also stably introduced into WT (DMPK CTG5) and DM1 (DMPK CTG50) fibroblasts, in an identical manner to the DM1 (DMPK CTG1000) cell line, generating additional tools for triaging hits according to their dependence upon CTG repeat length. Thus we have developed an ideal system for conducting HTS to better understand and treat DM1, a complex multi-systemic genetic disease.
ACKNOWLEDGEMENTS
The authors are grateful to Paul Anderson, Jeff Janes, Jeremy To and Vy Trinh for technical support, and Venkat Reddy for reagents. Thanks to Don MacKenzie, Karla Blonsky and Ian Atkinson for helpful discussion throughout the project, and Annie Mak for manuscript review. This work was supported by The Marigold Foundation.