- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Targets in Epigenetics: Inhibiting the Methyl Writers of the Histone Code
Julianne M Yost#, Ilia Korboukh#, Feng Liu#, Cen Gao#, Jian Jin*
Abstract
Growing evidence suggests that protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs) are associated with the development of various human diseases, including cancer, inflammation, and psychiatric disorders. Given the significant role of these proteins in human disease, efforts to discover selective small-molecule inhibitors of these enzymes are quickly gaining momentum. In this review, we focus on the recent progress in the discovery of selective PKMT and PRMT inhibitors. A future perspective on developing methyltransferase inhibitors is also offered.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 5
Issue: Suppl 1
First Page: 72
Last Page: 84
Publisher Id: CCGTM-5-72
DOI: 10.2174/1875397301005010072
Article History:
Received Date: 1/2/2011Revision Received Date: 11/7/2011
Acceptance Date: 18/7/2011
Electronic publication date: 22/8/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/ which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Center for Integrative Chemical Biology and Drug Discovery, Division of Medicinal Chemistry and Natural Products, UNC Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Tel: 919-843-8459; Fax: 919-843-8465; E-mail: jianjin@unc.edu# These authors contributed equally to this review.
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 1-2-2011 |
Original Manuscript | Targets in Epigenetics: Inhibiting the Methyl Writers of the Histone Code | |
1. INTRODUCTION
Although all nucleated cells contain the same genetic information, multicellular organisms have evolved elaborate mechanisms that enable differential and cell-type specific gene expression. Epigenetics refers to the heritable changes that control how the genome is accessed in different cell types during embryonic development and cellular differentiation [1Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics Genes Dev 2009; 23: 781-3.]. This capability permits specialization of function between cells without altering the DNA sequence. The cellular machinery that creates these heritable changes has been the subject of intense scientific investigation over the last decade [2Bernstein BE, Meissner A, Lander ES. The mammalian epigenome Cell 2007; 128: 669-81.].
The epigenome is a group of proteins that regulate gene expression and is responsible for cell differentiation [2Bernstein BE, Meissner A, Lander ES. The mammalian epigenome Cell 2007; 128: 669-81.]. The template upon which the epigenome is written is chromatin – the complex of DNA, histones and other structural proteins that efficiently package the genome in an appropriately accessible state within each cell [3Li B, Carey M, Workman JL. The role of chromatin during transcription Cell 2007; 128: 707-19., 4Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair Cell 2007; 128: 721-33.]. Regulation of chromatin state plays an important role in the fields of epigenetics, oncology, developmental biology, stem cell fate and regenerative medicine. The state of chromatin, and thus access to the genetic code, is mainly regulated by reversible post-translational modifications (PTMs) to DNA or histone proteins and the recognition of these marks by other proteins and protein complexes. Histones are subject to various kinds of PTMs, including methylation, acetylation, phosphorylation, sumoylation, ubiquitination, and glycosylation [5Kouzarides T. Chromatin modifications and their function Cell 2007; 128: 693-705., 6Gelato KA, Fischle W. Role of histone modifications in defining chromatin structure and function Biol Chem 2008; 389: 353-63.]. The addition, interpretation and removal of these PTM marks are crucial to gene transcription. Proteins that are directly involved in PTMs are often classified into three functional families: the enzymes that produce these modifications (the ‘writers’), the proteins that recognize them (the ‘readers’), and the enzymes that remove them (the ‘erasers’). Given the wide-spread importance of chromatin regulation in cell biology, the ‘writers’, ‘readers’, and ‘erasers’ are critical targets for manipulation to further understand the histone code [7Jenuwein T, Allis CD. Translating the histone code Science 2001; 293: 1074-80., 8Strahl BD, Allis CD. The language of covalent histone modifications Nature 2000; 403: 41-5.] and the role it plays in the progression of human disease.
Histone methylation, which mainly occurs at lysine and arginine residues located on the N-terminal tails of the core histones, is one of the most studied PTM marks [9Martin C, Zhang Y. The diverse functions of histone lysine methylation Nat Rev Mol Cell Biol 2005; 6: 838-49.]. Protein lysine methyltransferases (PKMTs) and protein arginine methyltransferases (PRMTs) are two of the writers responsible for adding the methyl marks to histones [10Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications Biochim Biophys Acta 2009; 1789: 45-57.]. Recently, it has also been shown that these writers are capable of methylating other functionally important proteins, including p53, ERα, pRb, TAF10, and HIV tat [11Jansson M, Durant ST, Cho E-C, et al. Arginine methylation regulates the p53 response Nat Cell Biol 2008; 10: 1431-9.-14Huang J, Berger SL. The emerging field of dynamic lysine methylation of non-histone proteins Curr Opin Genet Dev 2008; 18: 152-8.]. Both in vitro and cell-based studies have suggested that methyltransferases are responsible for methylating only specific residues, which is referred to as substrate selectivity. Moreover, these writers only methylate the residue to a specific methylation level, which is known as product specificity. For example, lysine residues can be mono-, di- or trimethylated (Scheme 1A), while arginines can be mono- or dimethylated in either a symmetric (sDMA) or asymmetric (aDMA) manner (Scheme 1B). Table 1 lists known histone methyl marks, their corresponding methyltransferases, and potential function and links to disease. Several experimental and theoretical groups have recently investigated the detailed catalytic mechanism, especially in regard to how writers achieve their substrate selectivity and product specificity [15Couture JF, Collazo E, Hauk G, Trievel RC. Structural basis for the methylation site specificity of SET7/9 Nat Struct Mol Biol 2006; 13: 140-6.-19Chu Y, Xu Q, Guo H. Understanding energetic origins of product specificity of SET8 from QM/MM free energy simulations: What causes the stop of methyl addition during histone lysine methylation? J Chem Theory Comput 2010; 6: 1380-9.]. These studies are not the subject of this review.
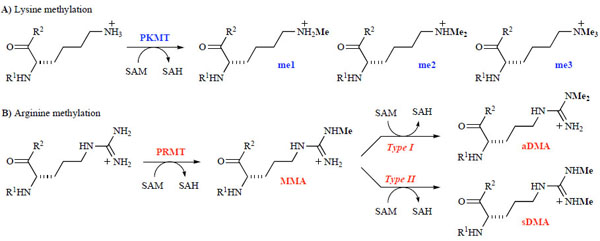 |
Scheme 1 SAM-dependent methylation of lysine (A) and arginine (B) residues. |

PKMTs and PRMTs have two binding pockets: the substrate binding pocket and a cofactor binding site [68Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570.]. Upon activation, the writer recruits the lysine or arginine residue of the histone tail to the substrate binding pocket and a universal methyl donor, S-5’-adenosyl-L-methionine (SAM), to the cofactor binding site [5Kouzarides T. Chromatin modifications and their function Cell 2007; 128: 693-705.]. As shown in Fig. (1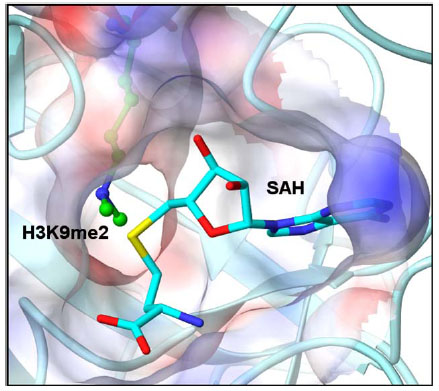 ), the substrate binding pocket and the cofactor binding site are joined by a narrow hydrophobic channel, just large enough to allow the terminal amine of the substrate to come within bonding distance of the cofactor. The terminal amine undergoes nucleophilic substitution with SAM, transferring the methyl group to the target residue, converting SAM to S-adenosyl-L-homocysteine (SAH) which is then released by the enzyme. In some cases, depending on the enzyme, this process can be repeated to add subsequent methyl groups to the lysine or arginine residue.
), the substrate binding pocket and the cofactor binding site are joined by a narrow hydrophobic channel, just large enough to allow the terminal amine of the substrate to come within bonding distance of the cofactor. The terminal amine undergoes nucleophilic substitution with SAM, transferring the methyl group to the target residue, converting SAM to S-adenosyl-L-homocysteine (SAH) which is then released by the enzyme. In some cases, depending on the enzyme, this process can be repeated to add subsequent methyl groups to the lysine or arginine residue.
 |
Fig. (1) GLP-H3 co-crystal structure (PDB: 2RFI, H3K9me2 is shown in green) with SAH (cyan) illustrating the hydrophobic channel connecting the cofactor binding site and the substrate binding pocket. |
Growing evidence suggests that PKMTs and PRMTs are associated with the development of various human diseases, including cancer, inflammation, and psychiatric disorders [69Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery Nat Rev Drug Discov 2009; 8: 724-32.-71Jones PA, Baylin SB. The epigenomics of cancer Cell 2007; 128: 683-92.]. For example, H3K9 methyltransferase G9a is overexpressed in various human cancers [26Huang J, Sengupta R, Espejo AB. p53 is regulated by the lysine demethylase LSD1 Nature 2007; 449: 105-8.-29Watanabe H, Soejima K, Yasuda H, et al. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells Cancer Cell Int 2008; 8: 15.] including leukemia, prostate carcinoma, hepatocellular carcinoma, and lung cancer and has been shown to play a role in mental retardation [72Schaefer A, Sampath SC, Intrator A, et al. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex Neuron 2009; 64: 678-91.], inflammation [33Li Y, Reddy MA, Miao F, et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-κB-dependent inflammatory genes: Relevance to diabetes and inflammation J Biol Chem 2008; 283: 26771-81.], drug addiction [73Maze I, Covington HE 3rd, Dietz DM, et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity Science 2010; 327: 213-6.] and HIV-1 latency maintenance [74Imai K, Togami H, Okamoto T. Involvement of histone H3 lysine 9 (H3K9) methyl transferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294 J Biol Chem 2010; 285: 16538-45.]. Another PKMT, DOT1L, can be recruited by AF10, an MLL (mixed lineage leukemia) or CALM (clathrin-assembly protein-like lymphoid–myeloid) fusion partner involved in acute myeloid leukemia [51Okada Y, Jiang Q, Lemieux M, Jeannotte L, Su L, Zhang Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L Nat Cell Biol 2006; 8: 1017-24., 52Okada Y, Feng Q, Lin Y, et al. hDOT1L links histone methylation to leukemogenesis Cell 2005; 121: 167-78.]. Direct fusion of DOT1L to MLL results in leukemic transformation in a DOT1L methyltransferase activity-dependent manner [52Okada Y, Feng Q, Lin Y, et al. hDOT1L links histone methylation to leukemogenesis Cell 2005; 121: 167-78.]. In addition, DOT1L contributes to CALM–AF10-mediated leukaemogenesis by methylation of H3K79 at the Hoxa5 gene [51Okada Y, Jiang Q, Lemieux M, Jeannotte L, Su L, Zhang Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L Nat Cell Biol 2006; 8: 1017-24.]. PRMT4 [better known as coactivator-associated arginine methyltransferase 1 (CARM1)] enhances transcriptional activation of nuclear receptors and aberrant expression of this methyltransferase has been linked to human breast and castration-resistant prostate cancers [60Majumder S, Liu Y, Ford OH, Mohler JL, Whang YE. Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability Prostate 2006; 66: 1292-301., 61Frietze S, Lupien M, Silver PA, Brown M. CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1 Cancer Res 2008; 68: 301-6.]. Given the significant role of these proteins in human disease, the discovery of potent, selective, and cell penetrant chemical probes of PKMTs and PRMTs is gaining momentum. In addition to providing the scientific community excellent tools for further studying chromatin remodeling and other PTMs, high quality well characterized chemical probes [75Frye SV. The art of the chemical probe Nat Chem Biol 2010; 6: 159-61.] can facilitate a smooth transition from target validation to a drug development effort. In this review, we will focus on the recent progress in the discovery of selective PKMT and PRMT inhibitors. A future perspective on developing methyltransferase inhibitors will also be discussed. Nucleoside-based inhibitors (analogs of cofactor SAM) such as sinefungin and methylthioadenosine and bisubstrate analogs containing structural components of both SAM and the substrate peptide will not be discussed in this review as these cofactor analogs are not selective in general [76Zheng YG, Wu J, Chen ZY, Goodman M. Chemical regulation of epigenetic modifications: Opportunities for new cancer therapy Med Res Rev 2008; 28: 645-87.-79Dowden J, Hong W, Parry RV, Pike RA, Ward SG. Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases Bioorg Med Chem Lett 2010; 20: 2103-5.].
2. PROTEIN LYSINE METHYLTRANSFERASES (PKMTs)
PKMTs can mono-, di-, and/or trimethylate lysine residues of various proteins including histones. Protein lysine methylation is involved in many biological processes, such as heterochromatin formation, gene expression, transcriptional regulation and X-chromosome inactivation [9Martin C, Zhang Y. The diverse functions of histone lysine methylation Nat Rev Mol Cell Biol 2005; 6: 838-49.]. Since the first PKMT was characterized in 2000 [80Rea S, Eisenhaber F, O'Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases Nature 2000; 406: 593-9.], more than 50 human PKMTs have been identified and over 10 high-resolution 3-dimensional (3-D) structures have been reported [5Kouzarides T. Chromatin modifications and their function Cell 2007; 128: 693-705., 9Martin C, Zhang Y. The diverse functions of histone lysine methylation Nat Rev Mol Cell Biol 2005; 6: 838-49., 69Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery Nat Rev Drug Discov 2009; 8: 724-32., 81Min JR, Feng Q, Li ZZ, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase Cell 2003; 112: 711-23.-85Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3.]. PKMTs are classified based on the presence or absence of the evolutionarily conserved SET domain, named after Drosophila Su(var)3-9 (suppressor of variegation 3-9), E(z) (enhancer of zeste), and trithorax [86Fog CK, Jensen KT, Lund AH. Chromatin-modifying proteins in cancer APMIS 2007; 115: 1060-89.]. The SET domain containing PKMTs are further categorized into five subfamilies named after their founding members: RIZ, SET1, SET2, SMYD3 and SUV39 [82Frye SV, Heightman T, Jin J. Targeting methyl lysine Annu Rep Med Chem 2010; 45: 329-43.].
A number of lysine methylation sites of H3 and H4 have been well characterized [87Dambacher S, Hahn M, Schotta G. Epigenetic regulation of development by histone lysine methylation Heredity 2010; 105: 24-37.]. Unlike acetylation which generally correlates with transcriptional activation [3Li B, Carey M, Workman JL. The role of chromatin during transcription Cell 2007; 128: 707-19.], histone lysine methylation can either activate or repress transcription depending on the site of methylation. For example, methylation at H3K4, H3K36 and H3K79 has been linked to actively transcribed genes, whereas methylated H3K9, H3K27 and H4K20 are considered repressive marks [88Martin C, Zhang Y. The diverse functions of histone lysine methylation Nat Rev Mol Cell Biol 2005; 6: 838-49.]. In addition to histones, other proteins have been identified as PKMT substrates [12Chuikov S, Kurash JK, Wilson JR, et al. Regulation of p53 activity through lysine methylation Nature 2004; 432: 353-60., 13Shi XB, Kachirskaia L, Yamaguchi H, et al. Modulation of p53 function by SET8-mediated methylation at lysine 382 Mol Cell 2007; 27: 636-46., 89Rathert P, Dhayalan A, Murakami M, et al. Protein lysine methyltransferase G9a acts on non-histone targets Nat Chem Biol 2008; 4: 344-6.-91Pagans S, Kauder SE, Kaehlcke K, et al. The cellular lysine methyltransferase Set7/9-KMT7 binds HIV-1 TAR RNA, monomethylates the viral transactivator Tat, and enhances HIV transcription Cell Host Microbe 2010; 7: 234-44.]. One such example, the tumor suppressor protein p53, implicated in over 50% of cancers [92Huang J, Dorsey J, Chuikov S, et al. G9a and Glp methylate lysine 373 in the tumor suppressor p53 J Biol Chem 2010; 285: 9636-41.], can be methylated by various PKMTs with contrasting effects depending on the site of lysine methylation. For instance, methylation of p53 on K370 by SMYD2 [51Okada Y, Jiang Q, Lemieux M, Jeannotte L, Su L, Zhang Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L Nat Cell Biol 2006; 8: 1017-24.] or on K382 by SETD8 [13Shi XB, Kachirskaia L, Yamaguchi H, et al. Modulation of p53 function by SET8-mediated methylation at lysine 382 Mol Cell 2007; 27: 636-46.] results in transcriptional repression, whereas methylation at K372 by SET7/9 leads to activation of transcription [12Chuikov S, Kurash JK, Wilson JR, et al. Regulation of p53 activity through lysine methylation Nature 2004; 432: 353-60.]. In addition, G9a (also known as EHMT2) and GLP (also known as EHMT1), which share 80% sequence homology in their respective SET domains [84Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7., 93Kubicek S, O'Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase Mol Cell 2007; 25: 473-81.], have been found to methylate p53 at K373, correlating with inactivation of p53 [92Huang J, Dorsey J, Chuikov S, et al. G9a and Glp methylate lysine 373 in the tumor suppressor p53 J Biol Chem 2010; 285: 9636-41.].
2.1. PKMT Inhibitors
In 2005, Greiner and co-workers screened a library of ca. 3,000 compounds using a standard radioactive filter-binding assay [94Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9 Nat Chem Biol 2005; 1: 143-5.] and identified chaetocin (Fig. 2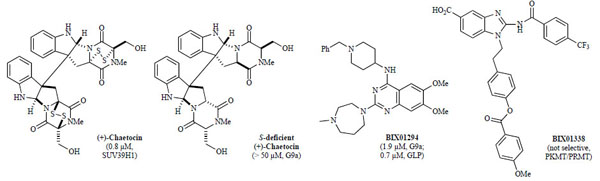 ), a fungal mycotoxin, as the first small-molecule inhibitor of recombinant Drosophila Su(var)3-9 (IC50 = 0.6 µM). Chaetocin was also found to inhibit H3K9 PKMT SUV39H1 (IC50 = 0.8 µM), the human orthologue of dSu(var)3-9. While chaetocin inhibited other H3K9 PKMTs, including Neurospora DIM5 (IC50 = 3.0 µM) and mouse G9a (IC50 = 2.5 µM), it was selective over non-H3K9 PKMTs, such as H3K27 PKMT dE(z) complex, H3K4 PKMT SET7/9, and H4K20 PKMT SETD8 [94Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9 Nat Chem Biol 2005; 1: 143-5.] (IC50 dE(z) complex > 90 µM; SET7/9 and SETD8 >180 µM). Furthermore, mechanistic studies characterized chaetocin as a SAM-competitive inhibitor, which remained active even after the disulfide bonds of chaetocin were reduced in the presence of increasing amounts of dithiothreitol (DTT) [94Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9 Nat Chem Biol 2005; 1: 143-5.]. Interestingly, a total synthesis report found both natural (+)- and synthetic (–)-chaetocin to be equipotent against G9a (IC50 = 2.4 and 1.7 µM, respectively) while the sulfur-deficient analogs were inactive (IC50 > 50 µM, Fig. 2) [95Iwasa E, Hamashima Y, Fujishiro S, et al. Total synthesis of (+)-chaetocin and its analogues: their histone methyltransferase G9a inhibitory activity J Am Chem Soc 2010; 132: 4078-9.]. Like other members of the epidithiodiketopiperazine (ETP) class [96Gardiner DM, Waring P, Howlett BJ. The epipolythiodioxopiperazine (ETP) class of fungal toxins: distribution, mode of action, functions and biosynthesis Microbiology 2005; 151: 1021-32.], chaetocin is cytotoxic, although dependent on initial cell density. Chaetocin-treated Drosophila SL-2 cells at an inhibitor concentration of 0.5 µM showed marked cellular reduction of di- and trimethylation levels of H3K9 with no apparent changes in the degree of methylation of other lysines (H3K27, H3K36, H3K79, and H3K4) [94Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9 Nat Chem Biol 2005; 1: 143-5.].
), a fungal mycotoxin, as the first small-molecule inhibitor of recombinant Drosophila Su(var)3-9 (IC50 = 0.6 µM). Chaetocin was also found to inhibit H3K9 PKMT SUV39H1 (IC50 = 0.8 µM), the human orthologue of dSu(var)3-9. While chaetocin inhibited other H3K9 PKMTs, including Neurospora DIM5 (IC50 = 3.0 µM) and mouse G9a (IC50 = 2.5 µM), it was selective over non-H3K9 PKMTs, such as H3K27 PKMT dE(z) complex, H3K4 PKMT SET7/9, and H4K20 PKMT SETD8 [94Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9 Nat Chem Biol 2005; 1: 143-5.] (IC50 dE(z) complex > 90 µM; SET7/9 and SETD8 >180 µM). Furthermore, mechanistic studies characterized chaetocin as a SAM-competitive inhibitor, which remained active even after the disulfide bonds of chaetocin were reduced in the presence of increasing amounts of dithiothreitol (DTT) [94Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9 Nat Chem Biol 2005; 1: 143-5.]. Interestingly, a total synthesis report found both natural (+)- and synthetic (–)-chaetocin to be equipotent against G9a (IC50 = 2.4 and 1.7 µM, respectively) while the sulfur-deficient analogs were inactive (IC50 > 50 µM, Fig. 2) [95Iwasa E, Hamashima Y, Fujishiro S, et al. Total synthesis of (+)-chaetocin and its analogues: their histone methyltransferase G9a inhibitory activity J Am Chem Soc 2010; 132: 4078-9.]. Like other members of the epidithiodiketopiperazine (ETP) class [96Gardiner DM, Waring P, Howlett BJ. The epipolythiodioxopiperazine (ETP) class of fungal toxins: distribution, mode of action, functions and biosynthesis Microbiology 2005; 151: 1021-32.], chaetocin is cytotoxic, although dependent on initial cell density. Chaetocin-treated Drosophila SL-2 cells at an inhibitor concentration of 0.5 µM showed marked cellular reduction of di- and trimethylation levels of H3K9 with no apparent changes in the degree of methylation of other lysines (H3K27, H3K36, H3K79, and H3K4) [94Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9 Nat Chem Biol 2005; 1: 143-5.].
 |
Fig. (2) Lysine methyltransferase inhibitors (IC50 values in parentheses with corresponding enzyme). |
A high throughput screen of ca. 125,000 compounds, preselected from the Boehringer Ingelheim (BI) compound collection, revealed BIX01294 (Fig. 2) as the first selective small-molecule inhibitor of G9a and GLP with low micromolar potency in vitro over other H3K9 PKMTs (SUV39H1 and SETDB1), H3K4 PKMT SET7/9, and arginine methyltransferase PRMT1, which all showed no inhibition at concentrations of 45 µ M [93Kubicek S, O'Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase Mol Cell 2007; 25: 473-81.]. Under linear assay conditions, BIX01294 inhibited G9a and GLP with IC50 values of 1.9 µ M and 0.7 µ M, respectively [84Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7.]. In cellular assays, BIX01294 was toxic at high concentrations (> 4.1 µ M). However, when cells were treated at an inhibitor concentration of 4.1 µ M, BIX01294 reduced H3K9me2 levels of bulk histones, while methylation levels of other known sites, including H3K27, H3K36, and H4K20, remained largely unchanged. Mechanistically, unlike chaetocin, BIX01294 did not inhibit G9a in a SAM-competitive manner but rather occupied the histone peptide binding pocket, as confirmed by the X-ray crystal structure of BIX01294 and GLP in the presence of SAH (Fig. 3A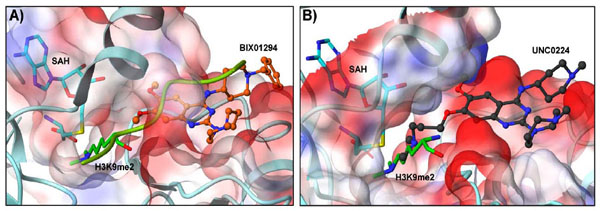 , PDB: 3FPD) [84Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7., 93Kubicek S, O'Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase Mol Cell 2007; 25: 473-81.]. Interestingly, the X-ray structure revealed that while BIX01294 did not bind in the SAM-binding site, it also did not interact with the lysine binding channel [84Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7.]. Through the same high-throughput screen as mentioned above, non-selective lysine and arginine methyltransferase inhibitors, such as BIX01338, were also discovered (Fig. 2) [93Kubicek S, O'Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase Mol Cell 2007; 25: 473-81.].
, PDB: 3FPD) [84Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7., 93Kubicek S, O'Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase Mol Cell 2007; 25: 473-81.]. Interestingly, the X-ray structure revealed that while BIX01294 did not bind in the SAM-binding site, it also did not interact with the lysine binding channel [84Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7.]. Through the same high-throughput screen as mentioned above, non-selective lysine and arginine methyltransferase inhibitors, such as BIX01338, were also discovered (Fig. 2) [93Kubicek S, O'Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase Mol Cell 2007; 25: 473-81.].

Structure-activity relationships (SAR) of the quinazoline scaffold exemplified by BIX01294 were investigated based on the reported X-ray structure of the GLP-BIX01294 complex (Fig. 3A). Tractable SAR were demonstrated for the 2- and 4-amino moieties [85Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3., 97Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines J Med Chem 2010; 53: 5844-7.]. To improve potency, the 7-methoxy moiety of the quinazoline template was explored in an attempt to design analogs that would interact with the lysine binding channel. These efforts resulted in the discovery of UNC0224 (Fig. 4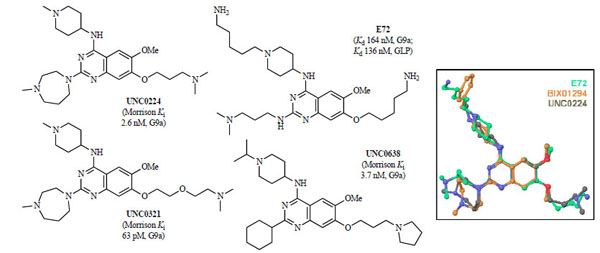 ) as a seven times more potent G9a inhibitor (IC50 = 15 nM) when compared to BIX01294 (IC50 = 106 nM) in the G9a ThioGlo assay [85Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3., 98Collazo E, Couture JF, Bulfer S, Trievel RC. A coupled fluorescent assay for histone methyltransferases Anal Biochem 2005; 342: 86-92.]. The higher potency of UNC0224 was confirmed by isothermal titration calorimetry (ITC) (Kd = 23 nM; BIX01294 Kd = 130 nM) [85Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3.]. Although UNC0224 was equipotent against GLP with an IC50 of 20 nM, it was more than 1,000-fold selective for G9a over other PKMTs, including SET7/9 and SETD8. In addition, UNC0224 was clean against a broad panel of G-protein coupled receptors, ion channels, and transporters [85Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3.]. A high resolution (1.7 Å) X-ray co-crystal structure of G9a and UNC0224 (PDB: 3K5K) was obtained, providing the first crystal structure of G9a in complex with a small molecule inhibitor (Fig. 3B). Indeed, the structure showed the 7-dimethylamino propoxy side chain of UNC0224 occupying the lysine binding channel of G9a, validating the design rationale for UNC0224 and providing an explanation for its higher potency [84Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7., 85Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3.]. However, the 7-alkoxy side chain did not completely occupy the lysine binding channel, and space remained for the channel to accommodate a longer side chain or larger amino-capping group. Therefore, additional SAR of the 7-alkoxy side chain of UNC0224 was investigated. These side chain optimization efforts led to the discovery of UNC0321 (Fig. 4), the most potent G9a inhibitor to date (IC50 = 6 nM, AlphaScreen; 9 nM, ThioGlo) [97Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines J Med Chem 2010; 53: 5844-7.]. Because UNC0321 likely reached the detection limits of the biochemical assays, Morrison Ki’s were determined using an endoproteinase-coupled microfluidic capillary electrophoresis (MCE) assay [99Wigle TJ, Provencher LM, Norris JL. Accessing protein methyltransferase and demethylase enzymology using microfluidic capillary electrophoresis Chem Biol 2010; 17: 695-704.]. UNC0321 (Morrison Ki = 63 pM) was about 40-fold more potent than UNC0224 (Morrison Ki = 2.6 nM) and 250-fold more potent than BIX01294 (Morrison Ki = 16 nM) [97Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines J Med Chem 2010; 53: 5844-7.]. While UNC0224 was equipotent against G9a and GLP, UNC0321 showed some selectivity for G9a (IC50 = 6 nM, AlphaScreen) over GLP (IC50 = 23 nM, AlphaScreen). In addition, UNC0321 was inactive (IC50 > 40 µM, ThioGlo) against other PKMTs, SET7/9 and SETD8, as well as PRMT3 [97Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines J Med Chem 2010; 53: 5844-7.]. While highly potent in biochemical assays, UNC0321 was less potent in cellular assays compared to BIX01294.
) as a seven times more potent G9a inhibitor (IC50 = 15 nM) when compared to BIX01294 (IC50 = 106 nM) in the G9a ThioGlo assay [85Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3., 98Collazo E, Couture JF, Bulfer S, Trievel RC. A coupled fluorescent assay for histone methyltransferases Anal Biochem 2005; 342: 86-92.]. The higher potency of UNC0224 was confirmed by isothermal titration calorimetry (ITC) (Kd = 23 nM; BIX01294 Kd = 130 nM) [85Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3.]. Although UNC0224 was equipotent against GLP with an IC50 of 20 nM, it was more than 1,000-fold selective for G9a over other PKMTs, including SET7/9 and SETD8. In addition, UNC0224 was clean against a broad panel of G-protein coupled receptors, ion channels, and transporters [85Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3.]. A high resolution (1.7 Å) X-ray co-crystal structure of G9a and UNC0224 (PDB: 3K5K) was obtained, providing the first crystal structure of G9a in complex with a small molecule inhibitor (Fig. 3B). Indeed, the structure showed the 7-dimethylamino propoxy side chain of UNC0224 occupying the lysine binding channel of G9a, validating the design rationale for UNC0224 and providing an explanation for its higher potency [84Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7., 85Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3.]. However, the 7-alkoxy side chain did not completely occupy the lysine binding channel, and space remained for the channel to accommodate a longer side chain or larger amino-capping group. Therefore, additional SAR of the 7-alkoxy side chain of UNC0224 was investigated. These side chain optimization efforts led to the discovery of UNC0321 (Fig. 4), the most potent G9a inhibitor to date (IC50 = 6 nM, AlphaScreen; 9 nM, ThioGlo) [97Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines J Med Chem 2010; 53: 5844-7.]. Because UNC0321 likely reached the detection limits of the biochemical assays, Morrison Ki’s were determined using an endoproteinase-coupled microfluidic capillary electrophoresis (MCE) assay [99Wigle TJ, Provencher LM, Norris JL. Accessing protein methyltransferase and demethylase enzymology using microfluidic capillary electrophoresis Chem Biol 2010; 17: 695-704.]. UNC0321 (Morrison Ki = 63 pM) was about 40-fold more potent than UNC0224 (Morrison Ki = 2.6 nM) and 250-fold more potent than BIX01294 (Morrison Ki = 16 nM) [97Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines J Med Chem 2010; 53: 5844-7.]. While UNC0224 was equipotent against G9a and GLP, UNC0321 showed some selectivity for G9a (IC50 = 6 nM, AlphaScreen) over GLP (IC50 = 23 nM, AlphaScreen). In addition, UNC0321 was inactive (IC50 > 40 µM, ThioGlo) against other PKMTs, SET7/9 and SETD8, as well as PRMT3 [97Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines J Med Chem 2010; 53: 5844-7.]. While highly potent in biochemical assays, UNC0321 was less potent in cellular assays compared to BIX01294.
 |
Fig. (4) Lysine methyltransferase inhibitors (Morrison Ki or Kd values in parentheses with corresponding enzyme); inset, E72 (green) super-imposed with BIX01294 (orange) and UNC0224 (grey). |
To improve cellular potency of this series, new analogs aimed at increasing lipophilicity, thus cell membrane permeability, while maintaining high in vitro potency were designed and synthesized. Among the newly synthesized compounds, UNC0638 (Fig. 4) had excellent in vitro potency (Morrison Ki G9a = 3.7 nM; Ki = 3.0 nM) and was > 100-fold selective over a wide range of epigenetic and non-epigenetic targets [100Vedadi M, Barsyte-Lovejoy D, Liu F, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells Nat Chem Biol 2011; 7: 566-74.]. Michaelis-Menten kinetics found UNC0638 was indeed competitive with the peptide substrate instead of the SAM cofactor. This mechanism of action was confirmed by X-ray crystal structure of the G9a-UNC0638-SAH complex (2.56 Å resolution, PDB: 3RJW) which showed UNC0638 occupying the substrate binding groove and lysine binding channel, not the SAM binding pocket – the same binding mode that was previously observed for UNC0224 (see Fig. 3B). More importantly, UNC0638, which possesses balanced in vitro potency and physicochemical properties aiding cell penetration, had excellent potency in cellular assays and low cell toxicity. UNC0638 treatment of a variety of cell lines resulted in reduction of global H3K9me2 levels equivalent to that observed for shRNA knockdown of G9a/GLP. It significantly reduced the H3K9me2 mark at the promoter of known G9a-regulated endogenous genes and did not reduce the H3K9me2 mark at the promoter of a non-G9a-regulated gene. In addition, UNC0638 significantly reduced the clonogenicity of MCF7 cells and disproportionately affected a number of genomic loci encoding microRNAs. In mouse embryonic stem (mES) cells, UNC0638 reactivated a retroviral reporter gene and G9a-silenced endogenous genes in a concentration dependent manner without promoting differentiation. Furthermore, UNC0638 significantly reduced H3K9me2 levels at the promoter of the retroviral long terminal repeat and G9a-regulated genes and indirectly induced DNA hypomethylation in mES cells [100Vedadi M, Barsyte-Lovejoy D, Liu F, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells Nat Chem Biol 2011; 7: 566-74.]. The combination of high potency, excellent selectivity, and robust on-target activities in cells makes UNC0638 a valuable tool for further investigating the biological function of G9a and GLP and their role in health and disease [101For the latest unpublished progress in this area, see: http://www.thesgc.org/chemical_probes/UNC0638/#overview ].
Chang and co-workers also designed and synthesized quinazoline analogs based on the previously reported GLP-BIX01294 X-ray co-crystal structure and discovered compound E72 (Fig. 4) as a potent G9a and GLP inhibitor (IC50 GLP = 100 nM) with binding affinities determined by ITC (Kd GLP = 136 nM; G9a = 164 nM) [102Chang Y, Ganesh T, Horton JR, et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases J Mol Biol 2010; 400: 1-7.]. A brief selectivity study showed E72 was inactive against H3K9 PKMT Suv39H2 with no inhibition at 5 µ M [102Chang Y, Ganesh T, Horton JR, et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases J Mol Biol 2010; 400: 1-7.]. The X-ray co-crystal structure of the GLP-E72 complex in the presence of SAH (2.19 Å, PDB: 3MO5) showed that E72 occupied both the surface of the peptide binding groove and the lysine binding channel, in an analogous manner to UNC0224 with G9a (see Fig. 4 inset for an overlay of E72 with BIX01294 and UNC0224) [102Chang Y, Ganesh T, Horton JR, et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases J Mol Biol 2010; 400: 1-7.]. In three separate cell types, E72 was significantly less toxic than BIX01294 at compound concentrations of 10 µ M and was able to reactivate K-ras-mediated epigenetic silencing of the Fas gene in NIH 3T3 cells [102Chang Y, Ganesh T, Horton JR, et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases J Mol Biol 2010; 400: 1-7.].
3. PROTEIN ARGININE METHYLTRANSFERASES (PRMTs)
Arginine residues of various histone and protein substrates can be mono- or dimethylated, and depending on the type of arginine methyltransferase, dimethlyation can occur in either a symmetric (one methyl group on each of the two terminal N atoms) or asymmetric (both methyl groups on the same terminal N atom) manner (Scheme 1B). These enzymes transfer a methyl group from SAM to a terminal (or ω) guanidino nitrogen of arginine, resulting in SAH and methylarginine. To date, about nine human PRMTs have been characterized and are mainly classified as type I or type II enzymes [59Di Lorenzo A, Bedford MT. Histone arginine methylation FEBS Lett 2010. DOI: 10.1016/j.febslet.2010.11.010]. Both types catalyze the formation of the monomethylarginine (MMA) intermediate. Type I enzymes (PRMT1–4, 6, and 8) then lead to the formation of asymmetric dimethylarginines (aDMA); whereas, type II enzymes (PRMT5, 7, and 9/FBXO11) form symmetrical dimethylarginine side chains (sDMA) [59Di Lorenzo A, Bedford MT. Histone arginine methylation FEBS Lett 2010. DOI: 10.1016/j.febslet.2010.11.010, 103Lee YH, Stallcup MR. Minireview: Protein arginine methylation of nonhistone proteins in transcriptional regulation Mol Endocrinol 2009; 23: 425-33., 104Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S. Protein arginine methyltransferases: Evolution and assessment of their pharmacological and therapeutic potential Pharmacol Ther 2007; 113: 50-87.]. In addition, PRMT7 exhibits type III enzymatic activity, the ability to selectively monomethylate certain substrates [59Di Lorenzo A, Bedford MT. Histone arginine methylation FEBS Lett 2010. DOI: 10.1016/j.febslet.2010.11.010, 105Miranda TB, Miranda M, Frankel A, Clarke S. PRMT7 is a member of the protein arginine methyltransferase family with a distinct substrate specificity J Biol Chem 2004; 279: 22902-7.]. Type IV activity has also been described, where the internal guanidino nitrogen atom is monomethylated, but this has only been documented in yeast [106Bedford MT. Arginine methylation at a glance J Cell Sci 2007; 120: 4243-6.]. Type I methyltransferase activity was recently demonstrated for PRMT2, albeit very weak [59Di Lorenzo A, Bedford MT. Histone arginine methylation FEBS Lett 2010. DOI: 10.1016/j.febslet.2010.11.010, 107Lakowski TM, Frankel A. Kinetic analysis of human protein arginine N-methyltransferase 2: formation of monomethyl- and asymmetric dimethyl-arginine residues on histone H4 Biochem J 2009; 421: 253-61.]. Also, while type II PRMT activity has been proposed for FLAG-PRMT9/hFBXO11 [108Cook JR, Lee JH, Yang ZH, et al. FBXO11/PRMT9, a new protein arginine methyltransferase, symmetrically dimethylates arginine residues Biochem Biophys Res Commun 2006; 342: 472-81.], none was detected with GST-purified hFBXO11 and GST-purified or HA-tagged DRE-1, the C. elegans ortholog [109Fielenbach N, Guardavaccaro D, Neubert K, et al. DRE-1: An evolutionarily conserved F box protein that regulates C-elegans developmental age Dev Cell 2007; 12: 443-55.]. Catalytic activity for PRMT9/4q31 has yet to be demonstrated [59Di Lorenzo A, Bedford MT. Histone arginine methylation FEBS Lett 2010. DOI: 10.1016/j.febslet.2010.11.010]. PRMTs usually methylate glycine- and arginine-rich patches (so-called GAR motifs) [106Bedford MT. Arginine methylation at a glance J Cell Sci 2007; 120: 4243-6., 110Pahlich S, Bschir K, Chiavi C, Belyanskaya L, Gehring H. Different methylation characteristics of protein arginine methyltransferase 1 and 3 toward the Ewing sarcoma protein and a peptide Proteins: Struct Funct Bioinform 2005; 61: 164-75.] except for PRMT4 (CARM1) which instead has an affinity toward proline, glycine, methionine, and arginine-rich sequences (PGM motifs) [111Cheng DH, Cote J, Shaaban S, Bedford MT. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing Mol Cell 2007; 25: 71-83., 112Bedford MT, Reed R, Leder P. WW domain-mediated interactions reveal a spliceosome-associated protein that binds a third class of proline-rich motif: The proline glycine and methionine-rich motif Proc Natl Acad Sci USA 1998; 95: 10602-7.]. PRMT5 has also shown an affinity for PGM motifs, in select cases [111Cheng DH, Cote J, Shaaban S, Bedford MT. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing Mol Cell 2007; 25: 71-83.].
PRMTs play a key role in transcriptional regulation, protein and RNA subcellular localization, RNA splicing, DNA damage repair and signal transduction [113Bedford MT, Richard S. Arginine methylation: An emerging regulator of protein function Mol Cell 2005; 18: 263-72.]. Arginine methylation has been implicated in the positive and negative regulation of transcription. At least two methyltransferases, PRMT1 and PRMT4 (CARM1), have been linked to transcriptional activation. PRMT1 serves as a diverse regulator of protein function, responsible for the bulk (ca. 85%) of total arginine methyltransferase activity, and has the ability to methylate H4R3. PRMT4 (CARM1) was identified as a steroid receptor coactivator and can catalyze the methylation of H3R2, H2R17, and H3R26. Certain transcription factors (NR, p53, YY1, NF-κB) recruit these enzymes to specific promoters where they activate transcription. In contrast, PRMT5 methylates H3R8 and H4R3 and serves as a strong repressor of numerous genes. For example, PRMT5 negatively regulates the expression of tumor suppressor genes ST7 and NM23 [63Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes Mol Cell Biol 2004; 24: 9630-45.].
3.1. PRMT Inhibitors
In 2004, Bedford and co-workers discovered the first non-nucleoside specific inhibitors of PRMTs using a random screening approach [114Cheng DH, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases J Biol Chem 2004; 279: 23892-9.]. A diverse library of 9,000 compounds from ChemBridge was screened and nine of these compounds were identified as low micromolar inhibitors (0.15–6.9 µM) of in vitro methylation of the RNA binding protein, Npl3p, by the yeast Hmt1p arginine methyltransferase. These hits, named arginine methyltransferase inhibitors (AMIs), also inhibited human PRMT1, a mammalian orthologue of Hmt1p, with potency ranging from 0.19 to 16.3 µM (Fig. 5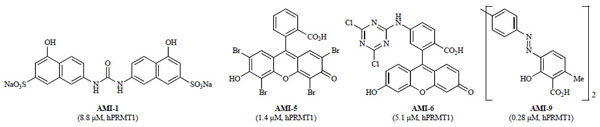 ) [114Cheng DH, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases J Biol Chem 2004; 279: 23892-9.].
) [114Cheng DH, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases J Biol Chem 2004; 279: 23892-9.].
 |
Fig. (5) Select examples of the first non-nucleoside arginine methyltransferase inhibitors (AMIs) identified from random screening (IC50 values in parentheses with corresponding enzyme). |
To determine specificities, the AMIs were screened against a panel of other type I PRMTs (PRMT3, 4, and 6). Although each of the nine AMIs initially inhibited all of the PRMTs tested, displaying no specificity for individual type I PRMTs [114Cheng DH, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases J Biol Chem 2004; 279: 23892-9.], a subsequent report found AMI-1 was selective over type II PRMT5 [115Bonham K, Hemmers S, Lim YH, Hill DM, Finn MG, Mowen KA. Effects of a novel arginine methyltransferase inhibitor on T-helper cell cytokine production FEBS J 2010; 277: 2096-108.]. Furthermore, AMI-1 and -6 were selective in vitro against several lysine methyltransferases including Suv39H1, Suv39H2, SET7/9, and DOT1. Cellular activity studies in HeLa cells showed AMI-1, but not -6, was able to inhibit hPRMT1, decreasing methylation levels of the GFP-Npl3 fusion [114Cheng DH, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases J Biol Chem 2004; 279: 23892-9.]. AMI-1 was also screened against sirtuin, a class III histone deacetylase (HDAC), due to the minor similarity of the binaphthylurea motif of AMI-1 to the known sirtuin inhibitor, suramin. AMI-1 inhibited sirtuin, albeit with decreased potency relative to suramin (IC50 on SIRT1: AMI-1, 32 µ M; suramin, 300 nM) [116Trapp J, Meier R, Hongwiset D, Kassack MU, Sippl W, Jung M. Structure-activity studies on suramin analogues as inhibitors of NAD(+)-dependent histone deacetylases (sirtuins) ChemMedChem 2007; 2: 1419-31.].
Several groups have since used the AMIs as leads for PRMT drug discovery. Using the dye-like core of AMI-5 and -6 as a lead scaffold, a number of analogs were synthesized [117Ragno R, Simeoni S, Castellano S, et al. Small molecule inhibitors of histone arginine methyltransferases: Homology modeling, molecular docking, binding mode analysis, and biological evaluations J Med Chem 2007; 50: 1241-53.]. The most potent of these analogs was compound 1 (IC50 hPRMT1 = 4.8 µ M), which was less potent than AMI-5 (Fig. 6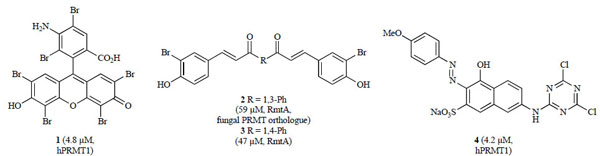 ). Cellular activity data was not reported [117Ragno R, Simeoni S, Castellano S, et al. Small molecule inhibitors of histone arginine methyltransferases: Homology modeling, molecular docking, binding mode analysis, and biological evaluations J Med Chem 2007; 50: 1241-53.]. The AMI-5 (eosin) scaffold was subsequently simplified to curcumin-like structures [118Balasubramanyam K, Varier RA, Altaf M, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription J Biol Chem 2004; 279: 51163-71.] containing bromo- and dibromophenol moieties [119Mai A, Cheng D, Bedford MT, et al. Epigenetic multiple ligands: Mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors J Med Chem 2008; 51: 2279-90.]. These analogs were screened against a panel of methyltransferases. Several compounds, including 2 and 3 (Fig. 6), were selective for PRMT4 (CARM1) over PRMT1 and SET7/9 at concentrations of 100 µ M [119Mai A, Cheng D, Bedford MT, et al. Epigenetic multiple ligands: Mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors J Med Chem 2008; 51: 2279-90.].
). Cellular activity data was not reported [117Ragno R, Simeoni S, Castellano S, et al. Small molecule inhibitors of histone arginine methyltransferases: Homology modeling, molecular docking, binding mode analysis, and biological evaluations J Med Chem 2007; 50: 1241-53.]. The AMI-5 (eosin) scaffold was subsequently simplified to curcumin-like structures [118Balasubramanyam K, Varier RA, Altaf M, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription J Biol Chem 2004; 279: 51163-71.] containing bromo- and dibromophenol moieties [119Mai A, Cheng D, Bedford MT, et al. Epigenetic multiple ligands: Mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors J Med Chem 2008; 51: 2279-90.]. These analogs were screened against a panel of methyltransferases. Several compounds, including 2 and 3 (Fig. 6), were selective for PRMT4 (CARM1) over PRMT1 and SET7/9 at concentrations of 100 µ M [119Mai A, Cheng D, Bedford MT, et al. Epigenetic multiple ligands: Mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors J Med Chem 2008; 51: 2279-90.].
 |
Fig. (6) Synthetic analogs of the original AMI series identified as PRMT inhibitors (IC50 values in parentheses with corresponding enzyme). |
In addition, Bonham and co-workers reported their efforts to generate a less polar version of AMI-1 while still maintaining PRMT potency [115Bonham K, Hemmers S, Lim YH, Hill DM, Finn MG, Mowen KA. Effects of a novel arginine methyltransferase inhibitor on T-helper cell cytokine production FEBS J 2010; 277: 2096-108.]. Using the aminonaphthol sulfonate scaffold of AMI-1 as the core, analogs were synthesized by adding structural attributes of AMI-6 and -9. The most potent hybrid, compound 4 (IC50 hPRMT1 = 4.2 µM; hPRMT4 = 2.6 µ M), contained both the dichlorotriazine group of AMI-6 and the azo moiety of AMI-9 (Fig. 6). Selectivity studies showed compound 4 inhibited both type I and type II PRMTs but was mostly inactive against lysine methyltransferase SET7/9. Since PRMTs have been shown to regulate T-helper cell activation and cytokine secretion [120Mowen KA, Schurter BT, Fathman JW, David M, Glimcher LH. Arginine methylation of NIP45 modulates cytokine gene expression in effector T lymphocytes Mol Cell 2004; 15: 559-71.-122Blanchet F, Cardona A, Letimier FA, Hershfield MS, Acuto O. CD28 costimulatory signal induces protein arginine methylation in T cells J Exp Med 2005; 202: 371-7.], the effect of compound 4 on cytokine expression was also examined. Indeed, compound 4 enhanced T-helper cell proliferation without affecting cell viability and decreased IFN-γ and IL-4 production of type I and type II T-helper cells, respectively, thereby interfering with IL-4 promoter activity and impairing the interaction between PRMT1 and NIP45 [115Bonham K, Hemmers S, Lim YH, Hill DM, Finn MG, Mowen KA. Effects of a novel arginine methyltransferase inhibitor on T-helper cell cytokine production FEBS J 2010; 277: 2096-108.].
A high throughput screening effort by Purandare and coworkers led to the identification of pyrazole amide 5 as an initial hit (Fig. 7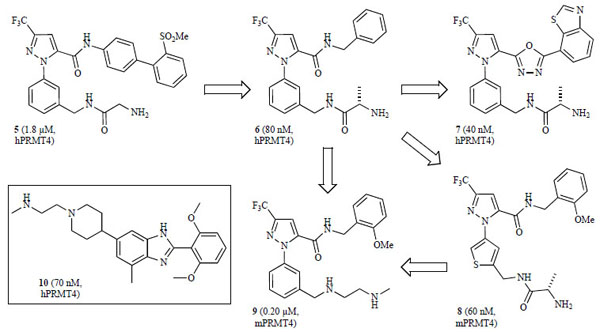 ), which after preliminary optimization gave compound 6 (IC50 hPRMT4 = 80 nM) as a potent and selective inhibitor of PRMT4 (CARM1) [123Purandare AV, Chen Z, Huynh T, et al. Pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2008; 18: 4438-1.], albeit poor permeability [parallel artificial membrane permeability assay (PAMPA)] and pharmacokinetic (PK) properties [124Huynh T, Chen Z, Pang S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 2924-7., 125Therrien E, Larouche G, Manku S, et al. 1,2-Diamines as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 6725-32.]. Further hit to lead optimization of this class by separate groups led to two potent derivatives: 7 and 8 (Fig. 7) with IC50’s < 100 nM, although the compounds were either not active in cellular assays or the data were not reported [124Huynh T, Chen Z, Pang S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 2924-7., 126Allan M, Manku S, Therrien E, et al. N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 1218-23.]. However, replacing the amide functionality with the 1,3,4-oxadiazole moiety in 7 improved membrane permeability (PAMPA) [124Huynh T, Chen Z, Pang S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 2924-7.]. Compounds 6 and 7 were found to be significantly less potent against other type I PRMTs (PRMT1 and 3, IC50 > 25 µM) [123Purandare AV, Chen Z, Huynh T, et al. Pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2008; 18: 4438-1., 124Huynh T, Chen Z, Pang S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 2924-7.] and 8 was selective against both PRMT1 and lysine methyltransferase SET7/9 (IC50 > 100 µ M) [126Allan M, Manku S, Therrien E, et al. N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 1218-23.]. A subsequent report identified compound 9 as a potent pyrazole derivative (IC50 mPRMT4 = 0.20 µ M, Fig. 7) with an improved PK profile in rats, although it did not show cellular activity when tested in the MTT cell viability assay [125Therrien E, Larouche G, Manku S, et al. 1,2-Diamines as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 6725-32.]. Concurrently, an additional screening effort by Wan and co-workers identified benzo[d]imidazole related analogs as hits against PRMT4 (CARM1) with low micromolar potency [127Wan HH, Huynh T, Pang SH, et al. Benzo[d]imidazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1)-hit to lead studies Bioorg Med Chem Lett 2009; 19: 5063-6.]. Hit to lead optimization and SAR exploration led to the most potent analog in this series, compound 10 (IC50 hPRMT4 = 70 nM, Fig. 7 inset), which was significantly less active against other type I PRMTs (PRMT1 and 3, IC50 > 25 µ M) [127Wan HH, Huynh T, Pang SH, et al. Benzo[d]imidazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1)-hit to lead studies Bioorg Med Chem Lett 2009; 19: 5063-6.].
), which after preliminary optimization gave compound 6 (IC50 hPRMT4 = 80 nM) as a potent and selective inhibitor of PRMT4 (CARM1) [123Purandare AV, Chen Z, Huynh T, et al. Pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2008; 18: 4438-1.], albeit poor permeability [parallel artificial membrane permeability assay (PAMPA)] and pharmacokinetic (PK) properties [124Huynh T, Chen Z, Pang S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 2924-7., 125Therrien E, Larouche G, Manku S, et al. 1,2-Diamines as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 6725-32.]. Further hit to lead optimization of this class by separate groups led to two potent derivatives: 7 and 8 (Fig. 7) with IC50’s < 100 nM, although the compounds were either not active in cellular assays or the data were not reported [124Huynh T, Chen Z, Pang S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 2924-7., 126Allan M, Manku S, Therrien E, et al. N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 1218-23.]. However, replacing the amide functionality with the 1,3,4-oxadiazole moiety in 7 improved membrane permeability (PAMPA) [124Huynh T, Chen Z, Pang S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 2924-7.]. Compounds 6 and 7 were found to be significantly less potent against other type I PRMTs (PRMT1 and 3, IC50 > 25 µM) [123Purandare AV, Chen Z, Huynh T, et al. Pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2008; 18: 4438-1., 124Huynh T, Chen Z, Pang S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 2924-7.] and 8 was selective against both PRMT1 and lysine methyltransferase SET7/9 (IC50 > 100 µ M) [126Allan M, Manku S, Therrien E, et al. N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 1218-23.]. A subsequent report identified compound 9 as a potent pyrazole derivative (IC50 mPRMT4 = 0.20 µ M, Fig. 7) with an improved PK profile in rats, although it did not show cellular activity when tested in the MTT cell viability assay [125Therrien E, Larouche G, Manku S, et al. 1,2-Diamines as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 6725-32.]. Concurrently, an additional screening effort by Wan and co-workers identified benzo[d]imidazole related analogs as hits against PRMT4 (CARM1) with low micromolar potency [127Wan HH, Huynh T, Pang SH, et al. Benzo[d]imidazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1)-hit to lead studies Bioorg Med Chem Lett 2009; 19: 5063-6.]. Hit to lead optimization and SAR exploration led to the most potent analog in this series, compound 10 (IC50 hPRMT4 = 70 nM, Fig. 7 inset), which was significantly less active against other type I PRMTs (PRMT1 and 3, IC50 > 25 µ M) [127Wan HH, Huynh T, Pang SH, et al. Benzo[d]imidazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1)-hit to lead studies Bioorg Med Chem Lett 2009; 19: 5063-6.].

The first target based virtual screening study for PRMTs was reported by Spannhoff and co-workers in 2007 [128Spannhoff A, Heinke R, Bauer I, et al. Target-based approach to inhibitors of histone arginine methyltransferases J Med Chem 2007; 50: 2319-5.-130Jung M. Epigenetic targets in drug discovery In: Sippl W, Jung M, Eds. Weinheim Wiley-VCH 2009; 251-68.]. In this study, a combination of molecular docking and pharamacophore-based filtering was used to virtually screen hPRMT1 and fungal RmtA, a PRMT1 homologue, against the NCI diversity subset consisting of 1990 compounds. Homology models of both enzymes were constructed from the known rat PRMT3 X-ray crystal structure (PDB: 1F3L). In order to target the histone binding pocket, SAH was included as part of the protein during docking. Compounds that were successfully docked were then tested in vitro against RmtA and recombinant hPRMT1. In this study, seven of the 36 virtual hits were able to inhibit RmtA and hPRMT1 with micromolar potency (IC50 hPRMT1 = 2–90 µ M) [128Spannhoff A, Heinke R, Bauer I, et al. Target-based approach to inhibitors of histone arginine methyltransferases J Med Chem 2007; 50: 2319-5.]. Hit validation was conducted in cancer cells using antibody-mediated detection of histone hypomethylation caused by inhibition of PRMT1. Two of the hits, allantodapsone and stilbamidine (Fig. 8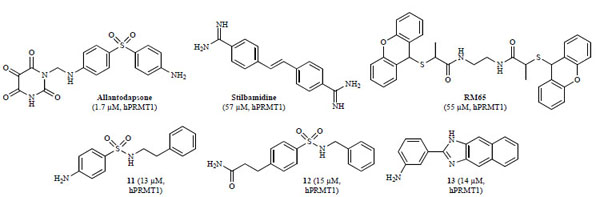 ), inhibited methylation at PRMT1 target H4R3 in a dose-dependent manner while having only a marginal effect on methylation levels of PKMT target H3K4. In a reporter gene functional assay with MCF7a cells, both inhibitors showed a dose-dependent reduction of estrogen receptor activation by estradiol. In addition, kinetic assays showed allantodapsone and stilbamidine did not inhibit RmtA in a SAM-competitive manner but were rather competitive with regard to the histone substrate, as expected [128Spannhoff A, Heinke R, Bauer I, et al. Target-based approach to inhibitors of histone arginine methyltransferases J Med Chem 2007; 50: 2319-5.].
), inhibited methylation at PRMT1 target H4R3 in a dose-dependent manner while having only a marginal effect on methylation levels of PKMT target H3K4. In a reporter gene functional assay with MCF7a cells, both inhibitors showed a dose-dependent reduction of estrogen receptor activation by estradiol. In addition, kinetic assays showed allantodapsone and stilbamidine did not inhibit RmtA in a SAM-competitive manner but were rather competitive with regard to the histone substrate, as expected [128Spannhoff A, Heinke R, Bauer I, et al. Target-based approach to inhibitors of histone arginine methyltransferases J Med Chem 2007; 50: 2319-5.].
 |
Fig. (8) PRMT inhibitors identified from virtual screens (IC50 values in parentheses with corresponding enzyme). |
Based on the success of the structure-based virtual screen to discover potent and cell-penetrant PRMT inhibitors, a larger virtual screening effort was conducted using the ChemBridge compound collection containing 328,000 compounds [129Bissinger EM, Heinke R, Sippl W, Jung M. Targeting epigenetic modifiers: Inhibitors of histone methyltransferases Med Chem Commun 2010; 1: 114-24.-131Heinke R, Spannhoff A, Meier R, et al. Virtual screening and biological characterization of novel histone arginine methyltransferase PRMT1 inhibitors ChemMedChem 2009; 4: 69-77.]. The database was first filtered using a simple pharmacophore search resulting in an abbreviated library of ca. 6200 compounds. These compounds were then docked into the substrate binding pocket of PRMT1, resulting in nine inhibitors of hPRMT1 with IC50 values ranging from 13 to 37 μM [131Heinke R, Spannhoff A, Meier R, et al. Virtual screening and biological characterization of novel histone arginine methyltransferase PRMT1 inhibitors ChemMedChem 2009; 4: 69-77.]. The three most potent compounds, 11–13, are represented in Fig. (8).
As an alternative approach for identifying new PRMT inhibitors, Spannhoff and co-workers performed a fragment-based virtual screen of ca. 900 fragment-like leads with molecular weights below 200 g/mol and identified an α-methylthioglycolic amide compound as a micromolar inhibitor of RmtA [129Bissinger EM, Heinke R, Sippl W, Jung M. Targeting epigenetic modifiers: Inhibitors of histone methyltransferases Med Chem Commun 2010; 1: 114-24., 130Jung M. Epigenetic targets in drug discovery In: Sippl W, Jung M, Eds. Weinheim Wiley-VCH 2009; 251-68., 132Spannhoff A, Machmur R, Heinke R, et al. A novel arginine methyltransferase inhibitor with cellular activity Bioorg Med Chem Lett 2007; 17: 4150-3.]. A subsequent structure similarity search led to the discovery of RM65 (Fig. 8) as an equipotent inhibitor of RmtA and hPRMT1 in vitro (IC50 hPRMT1 = 55 µ M). A cursory selectivity study found RM65 was not active against PKMT SET7/9 at a concentration of 50 µ M. In cancer cells, histone hypomethylation was observed at PRMT1 target H4R3. Additionally, while the virtual screen was conducted with fragments binding to the substrate binding pocket, docking studies have suggested a bisubstrate binding mode for RM65, targeting both the substrate and cofactor binding sites [132Spannhoff A, Machmur R, Heinke R, et al. A novel arginine methyltransferase inhibitor with cellular activity Bioorg Med Chem Lett 2007; 17: 4150-3.].
An alternative mechanism of inhibiting PRMT-mediated arginine methylation was recently reported. Rather than developing inhibitors of the enzyme active site, Feng and co-workers developed small molecules that target the substrate, blocking PTMs on H4 by H4-modifying enzymes [133Feng Y, Li MY, Wang BH, Zheng YG. Discovery and mechanistic study of a class of protein arginine methylation inhibitors J Med Chem 2010; 53: 6028-39.]. The naphthalene-sulfonate (NS) analogs were discovered through a similarity structure search of a weak hit identified from a virtual screen of the ChemBridge compound collection. The most potent derivative, NS-1 (Fig. 9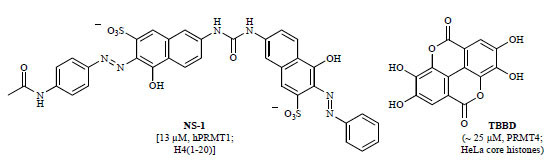 ), was initially characterized as a substrate-competitive PRMT1 inhibitor with micromolar potency (IC50 hPRMT1 = 13 µ M) [133Feng Y, Li MY, Wang BH, Zheng YG. Discovery and mechanistic study of a class of protein arginine methylation inhibitors J Med Chem 2010; 53: 6028-39.]. However, after a variety of kinetic and biophysical studies, NS-1 and similar structural analogs including AMI-1 were found to interact directly with the substrate, not the enzyme, blocking PRMT1-mediated arginine methylation. In a selectivity study, NS-1 was found to be significantly less potent against PRMT4 (CARM1) (IC50 mPRMT4 ~ 2 mM) [133Feng Y, Li MY, Wang BH, Zheng YG. Discovery and mechanistic study of a class of protein arginine methylation inhibitors J Med Chem 2010; 53: 6028-39.]. Unlike PRMT1, PRMT4 (CARM1) targets H3 and does not methylate GAR sequences. In addition, Selvi and co-workers isolated the small molecule TBBD (ellagic acid) from pomegranate extract and found it inhibited the methyltransferase activity of PRMT4 (CARM1) in a substrate-targeting manner, selectively blocking methylation of H3R17 (Fig. 9) [134Selvi BR, Batta K, Kishore AH, et al. Identification of a novel inhibitor of coactivator-associated arginine methyltransferase 1 (CARM1)-mediated methylation of histone H3 Arg-17 J Biol Chem 2010; 285: 7143-52.]. These and other substrate sequence-specific inhibitors may emerge as useful tools for mechanistic study of arginine methylation and other epigenetic modifications.
), was initially characterized as a substrate-competitive PRMT1 inhibitor with micromolar potency (IC50 hPRMT1 = 13 µ M) [133Feng Y, Li MY, Wang BH, Zheng YG. Discovery and mechanistic study of a class of protein arginine methylation inhibitors J Med Chem 2010; 53: 6028-39.]. However, after a variety of kinetic and biophysical studies, NS-1 and similar structural analogs including AMI-1 were found to interact directly with the substrate, not the enzyme, blocking PRMT1-mediated arginine methylation. In a selectivity study, NS-1 was found to be significantly less potent against PRMT4 (CARM1) (IC50 mPRMT4 ~ 2 mM) [133Feng Y, Li MY, Wang BH, Zheng YG. Discovery and mechanistic study of a class of protein arginine methylation inhibitors J Med Chem 2010; 53: 6028-39.]. Unlike PRMT1, PRMT4 (CARM1) targets H3 and does not methylate GAR sequences. In addition, Selvi and co-workers isolated the small molecule TBBD (ellagic acid) from pomegranate extract and found it inhibited the methyltransferase activity of PRMT4 (CARM1) in a substrate-targeting manner, selectively blocking methylation of H3R17 (Fig. 9) [134Selvi BR, Batta K, Kishore AH, et al. Identification of a novel inhibitor of coactivator-associated arginine methyltransferase 1 (CARM1)-mediated methylation of histone H3 Arg-17 J Biol Chem 2010; 285: 7143-52.]. These and other substrate sequence-specific inhibitors may emerge as useful tools for mechanistic study of arginine methylation and other epigenetic modifications.
 |
Fig. (9) Substrate-targeting PRMT inhibitors (IC50 values in parentheses with corresponding enzyme and substrate). |
4. FUTURE DIRECTIONS IN DRUG DISCOVERY
The interest in epigenetic misregulation from the scientific community over the past decade is bound to increase in the future as more scientists realize its profound effect on genome function [135Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery Nat Rev Drug Discov 2009; 8: 724-32.]. Understanding the mechanisms behind cell differentiation and gaining the ability to control them by inhibiting chromatin modifying enzymes holds much promise for combating human disease. For example, it is now understood that epigenetic changes work in concert with genetic changes to promote oncogenesis. Recently, viability of this approach has been confirmed by promising cancer therapy clinical data [136Pollock RM, Richon VM. Epigenetic approaches to cancer therapy Drug Discov Today: Ther Strategies 2009; 6: 71-9.]. These discoveries drive the interest in solving the mystery behind cell differentiation and deciphering the histone code.
As new inhibitors for various methyltransferases emerge, mechanisms by which PTM marks are made and their impact on nucleosome structure and remodeling are becoming better understood. Thus far, the histone code has proven to be a significantly more complex entity than for example, the genetic code, where a direct mapping between codons and amino acids exists [137Turanov AA, Lobanov AV, Fomenko DE, et al. Genetic code supports targeted insertion of two amino acids by one codon Science 2009; 323: 259-61.]. Certain correlations between specific PTMs of the histones and individual processes have been identified, inter alia, methylation of H3K4, H3K36 and H3K79 has been linked to activation of transcription, H3K79 and H4K20 to DNA repair, and H3K9 and H3K27 have been correlated to transcription repression [138Kouzarides T, Berger SL. Epigenetics. Cold Spring Harbor: Cold Spring Harbor Laboratory Press 2007; pp. 191-209.]. Interestingly, these correlations are not always conserved amongst species and organisms [139Garcia BA, Hake SB, Diaz RL, et al. Organismal differences in post-translational modifications in histones H3 and H4 J Biol Chem 2007; 282: 7641-55.].
The discovery effort is quickly gaining momentum and several potent, selective inhibitors of methyltransferases have already been identified vide supra. Future efforts should focus on continued discovery of potent, selective, and cell penetrant inhibitors. A number of traditional techniques have been employed in the discovery of the existing inhibitors. High throughput screening (HTS) and virtual screening (VS) are responsible for identifying most of these inhibitors. As the number of available HTS compatible biological assays increases, these approaches are likely to continue to generate structurally diverse hits as good starting points for medicinal chemistry optimization.
Increased availability of high resolution 3-dimensional (3D) structures of methyltransferases in apo- and holo- states provides medicinal and computational chemists a unique opportunity to design and synthesize novel inhibitors. Reviewing the existing crystal structure data, two distinct cofactor conformations can be noted for PKMTs and PRMTs. Fig. (10 ) shows the superimposition of the cofactor conformations extracted from the currently available crystal structures. Amongst the PKMTs, with the exception of DOT1L, the cofactor (SAM or SAH) adopts a highly conserved conformation, sometimes referred to as the “U shape” (Fig. 10A). On the other hand, a different and a more extended cofactor conformation is observed in four of the PRMTs, as well as for DOT1L (Fig. 10B). This consistent cofactor conformation may provide a molecular basis for ligand-based rational design as well as for pharmacophore-based screening. Although inhibitor selectivity could be a potential issue for this target class approach [140Gileadi O, Knapp S, Lee WH, et al. The scientific impact of the Structural Genomics Consortium: a protein family and ligand-centered approach to medically-relevant human proteins J Struct Funct Genomics 2007; 8: 107-9., 141Weigelt J, McBroom-Cerajewski LD, Schapira M, Zhao Y, Arrowsmith CH. Structural genomics and drug discovery: all in the family Curr Opin Chem Biol 2008; 12: 32-9.], one may reasonably expect that a high degree of selectivity can be achieved by targeting diversified residues in the binding pockets based on what we have learned from the discovery of highly selective kinase inhibitors. Drawing upon the analogy between SAM/methyltransferase and the ATP/kinase relationships, many of the techniques previously applied to studying kinases could very well be carried over to the investigation of methyltransferases [135Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery Nat Rev Drug Discov 2009; 8: 724-32.]. To date, few cofactor-competitive inhibitors have been reported. Thus, progress on a systematic approach to discovering SAM-competitive inhibitors is keenly awaited.
) shows the superimposition of the cofactor conformations extracted from the currently available crystal structures. Amongst the PKMTs, with the exception of DOT1L, the cofactor (SAM or SAH) adopts a highly conserved conformation, sometimes referred to as the “U shape” (Fig. 10A). On the other hand, a different and a more extended cofactor conformation is observed in four of the PRMTs, as well as for DOT1L (Fig. 10B). This consistent cofactor conformation may provide a molecular basis for ligand-based rational design as well as for pharmacophore-based screening. Although inhibitor selectivity could be a potential issue for this target class approach [140Gileadi O, Knapp S, Lee WH, et al. The scientific impact of the Structural Genomics Consortium: a protein family and ligand-centered approach to medically-relevant human proteins J Struct Funct Genomics 2007; 8: 107-9., 141Weigelt J, McBroom-Cerajewski LD, Schapira M, Zhao Y, Arrowsmith CH. Structural genomics and drug discovery: all in the family Curr Opin Chem Biol 2008; 12: 32-9.], one may reasonably expect that a high degree of selectivity can be achieved by targeting diversified residues in the binding pockets based on what we have learned from the discovery of highly selective kinase inhibitors. Drawing upon the analogy between SAM/methyltransferase and the ATP/kinase relationships, many of the techniques previously applied to studying kinases could very well be carried over to the investigation of methyltransferases [135Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery Nat Rev Drug Discov 2009; 8: 724-32.]. To date, few cofactor-competitive inhibitors have been reported. Thus, progress on a systematic approach to discovering SAM-competitive inhibitors is keenly awaited.

In addition, a fragment based drug discovery approach [142Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned Nat Rev Drug Discov 2007; 6: 211-9., 143Everts S. Piece by piece: More and more companies are using fragment-based lead design as a drug discovery strategy Chem Eng News 2008; 86: 15-23.], where weakly binding fragments are identified and then grown or combined to produce higher affinity molecules, might be employed in discovering novel methyltransferase inhibitors. Taking note of the close proximity observed between the cofactor and substrate binding pockets, one might envision designing a ligand that occupies both binding sites. Finally, molecules that interact with an allosteric site may yield quality methyltransferase inhibitors.
The scientific community eagerly awaits the discovery of more, high quality, well characterized chemical probes [75Frye SV. The art of the chemical probe Nat Chem Biol 2010; 6: 159-61.] of PKMTs and PRMTs. A ‘tool-kit’ of such chemical probes would permit biological hypotheses to be tested in cell-based and animal models of human biology and disease with high confidence, thus providing the biomedical community with excellent tools for further investigating the biology of methyltransferases and their role in chromatin remodeling and other PTMs.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGEMENTS
We thank the Carolina Partnership and the University Cancer Research Fund (UCRF) from the University of North Carolina at Chapel Hill for financial support.
REFERENCES
| [1] | Berger SL, Kouzarides T, Shiekhattar R, Shilatifard A. An operational definition of epigenetics Genes Dev 2009; 23: 781-3. |
| [2] | Bernstein BE, Meissner A, Lander ES. The mammalian epigenome Cell 2007; 128: 669-81. |
| [3] | Li B, Carey M, Workman JL. The role of chromatin during transcription Cell 2007; 128: 707-19. |
| [4] | Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair Cell 2007; 128: 721-33. |
| [5] | Kouzarides T. Chromatin modifications and their function Cell 2007; 128: 693-705. |
| [6] | Gelato KA, Fischle W. Role of histone modifications in defining chromatin structure and function Biol Chem 2008; 389: 353-63. |
| [7] | Jenuwein T, Allis CD. Translating the histone code Science 2001; 293: 1074-80. |
| [8] | Strahl BD, Allis CD. The language of covalent histone modifications Nature 2000; 403: 41-5. |
| [9] | Martin C, Zhang Y. The diverse functions of histone lysine methylation Nat Rev Mol Cell Biol 2005; 6: 838-49. |
| [10] | Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications Biochim Biophys Acta 2009; 1789: 45-57. |
| [11] | Jansson M, Durant ST, Cho E-C, et al. Arginine methylation regulates the p53 response Nat Cell Biol 2008; 10: 1431-9. |
| [12] | Chuikov S, Kurash JK, Wilson JR, et al. Regulation of p53 activity through lysine methylation Nature 2004; 432: 353-60. |
| [13] | Shi XB, Kachirskaia L, Yamaguchi H, et al. Modulation of p53 function by SET8-mediated methylation at lysine 382 Mol Cell 2007; 27: 636-46. |
| [14] | Huang J, Berger SL. The emerging field of dynamic lysine methylation of non-histone proteins Curr Opin Genet Dev 2008; 18: 152-8. |
| [15] | Couture JF, Collazo E, Hauk G, Trievel RC. Structural basis for the methylation site specificity of SET7/9 Nat Struct Mol Biol 2006; 13: 140-6. |
| [16] | Couture J-F, Dirk LMA, Brunzelle JS, Houtz RL, Trievel RC. Structural origins for the product specificity of SET domain protein methyltransferases Proc Natl Acad Sci USA 2008; 105: 20659-64. |
| [17] | Hu P, Zhang Y. Catalytic mechanism and product specificity of the histone lysine methyltransferase SET7/9: An ab initio QM/MM-FE study with multiple initial structures J Am Chem Soc 2006; 128: 1272-8. |
| [18] | Hu P, Wang S, Zhang Y. How do SET-domain protein lysine methyltransferases achieve the methylation state specificity? Revisited by ab initio QM/MM molecular dynamics simulations J Am Chem Soc 2008; 130: 3806-13. |
| [19] | Chu Y, Xu Q, Guo H. Understanding energetic origins of product specificity of SET8 from QM/MM free energy simulations: What causes the stop of methyl addition during histone lysine methylation? J Chem Theory Comput 2010; 6: 1380-9. |
| [20] | Kang MY, Lee BB, Kim Y-H, et al. Association of the SUV39H1 histone methyltransferase with the DNA methyltransferase 1 at mRNA expression level in primary colorectal cancer Int J Cancer 2007; 121: 2192-7. |
| [21] | Dodge JE, Kang YK, Beppu H, Lei H, Li E. Histone H3-K9 methyltransferase ESET is essential for early development Mol Cell Biol 2004; 24: 2478-86. |
| [22] | Tryndyak VP, Kovalchuk O, Pogribny IP. Loss of DNA methylation and histone H4 lysine 20 trimethylation in human breast cancer cells is associated with aberrant expression of DNA methyltransferase 1, Suv4-20h2 histone methyltransferase and methyl-binding proteins Cancer Biol Ther 2006; 5: 65-70. |
| [23] | Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics Mutat Res 2008; 647: 21-9. |
| [24] | Yap DB, Chu J, Berg T, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation Blood 2011; 117: 2451-9. |
| [25] | Sneeringer CJ, Scott MP, Kuntz KW, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas Proc Natl Acad Sci USA 2010; 107: 20980-5. |
| [26] | Huang J, Sengupta R, Espejo AB. p53 is regulated by the lysine demethylase LSD1 Nature 2007; 449: 105-8. |
| [27] | Kondo Y, Shen L, Suzuki S, et al. Alterations of DNA methylation and histone modifications contribute to gene silencing in hepatocellular carcinomas Hepatol Res 2007; 37: 974-83. |
| [28] | Kondo Y, Shen L, Ahmed S. Downregulation of histone H3 lysine 9 methyltransferase G9a induces centrosome disruption and chromosome instability in cancer cells PLoS One 2008; 3: e2037. |
| [29] | Watanabe H, Soejima K, Yasuda H, et al. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells Cancer Cell Int 2008; 8: 15. |
| [30] | Aniello F, Colella G, Muscariello G, et al. Expression of four histone lysine-methyltransferases in parotid gland tumors Anticancer Res 2006; 26: 2063-7. |
| [31] | Li HW, Rauch T, Chen ZX, Szabo PE, Riggs AD, Pfeifer GP. The histone methyltransferase SETDB1 and the DNA methyltransferase DNMT3A interact directly and localize to promoters silenced in cancer cells J Biol Chem 2006; 281: 19489-500. |
| [32] | Kurash JK, Lei H, Shen Q, et al. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo Mol Cell 2008; 29: 392-400. |
| [33] | Li Y, Reddy MA, Miao F, et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-κB-dependent inflammatory genes: Relevance to diabetes and inflammation J Biol Chem 2008; 283: 26771-81. |
| [34] | Rice JC, Nishioka K, Sarma K, Steward R, Reinberg D, Allis CD. Mitotic-specific methylation of histone H4 Lys 20 follows increased PR-Set7 expression and its localization to mitotic chromosomes Genes Dev 2002; 16: 2225-30. |
| [35] | Karachentsev D, Sarma K, Reinberg D, Steward R. PR-Set7-dependent methylation of histone H4 Lys 20 functions in repression of gene expression and is essential for mitosis Genes Dev 2005; 19: 431-5. |
| [36] | Shi X, Kachirskaia I, Yamaguchi H, et al. Modulation of p53 function by SET8-mediated methylation at lysine 382 Mol Cell 2007; 27: 636-46. |
| [37] | McCarthy N. Leukaemia: MLL makes friends and influences Nat Rev Cancer 2010; 10: 529. |
| [38] | Milne TA, Kim J, Wang GG, et al. Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis Mol Cell 2010; 38: 853-63. |
| [39] | Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development Nat Rev Cancer 2007; 7: 823-33. |
| [40] | Dalgliesh GL, Furge K, Greenman C, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes Nature 2010; 463: 360-. |
| [41] | Paulussen ADC, Stegmann APA, Blok MJ, et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome Hum Mutat DOI: 10.1002/humu.21416: Available from: http://dx.doi.org/10.1002/humu.21416 |
| [42] | Lee S, Lee DK, Dou YL, et al. Coactivator as a target gene specificity determinant for histone H3 lysine 4 methyltransferases Proc Natl Acad Sci USA 2006; 103: 15392-7. |
| [43] | Saigo K, Yoshida K, Ikeda R, et al. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma Hum Mutat 2008; 29: 703-8. |
| [44] | Huang J, Perez-Burgos L, Placek BJ, et al. Repression of p53 activity by Smyd2-mediated methylation Nature 2006; 444: 629-32. |
| [45] | Hamamoto R, Furukawa Y, Morita M, et al. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells Nat Cell Biol 2004; 6: 731-40. |
| [46] | Hamamoto R, Silva FP, Tsuge M, et al. Enhanced SMYD3 expression is essential for the growth of breast cancer cells Cancer Sci 2006; 97: 113-8. |
| [47] | Rio M, Clech L, Amiel J, et al. Spectrum of NSD1 mutations in Sotos and Weaver syndromes J Med Genet 2003; 40: 436-0. |
| [48] | Stec I, Wright TJ, van Ommen G-JB, et al. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(1;14) multiple myeloma Hum Mol Genet 1998; 7: 1071-82. |
| [49] | Rosati R, La Starza R, Veronese A, et al. NUP98 is fused to the NSD3 gene in acute myeloid leukemia associated with t(8;11)(p11.2;p15) Blood 2002; 99: 3857-60. |
| [50] | Angrand P-O, Apiou F, Stewart AF, Dutrillaux B, Losson R, Chambon P. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines Genomics 2001; 74: 79-88. |
| [51] | Okada Y, Jiang Q, Lemieux M, Jeannotte L, Su L, Zhang Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L Nat Cell Biol 2006; 8: 1017-24. |
| [52] | Okada Y, Feng Q, Lin Y, et al. hDOT1L links histone methylation to leukemogenesis Cell 2005; 121: 167-78. |
| [53] | Scorilas A, Black MH, Talieri M, Diamandis EP. Genomic organization, physical mapping, and expression analysis of the human protein arginine methyltransferase 1 gene Biochem Biophys Res Commun 2000; 278: 349-59. |
| [54] | Goulet I, Gauvin G, Boisvenue S, Cote J. Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization J Biol Chem 2007; 282: 33009-21. |
| [55] | Mathioudaki K, Papadokostopoulou A, Scorilas A, Xynopoulos D, Agnanti N, Talieri M. The PRMT1 gene expression pattern in colon cancer Br J Cancer 2008; 99: 2094-9. |
| [56] | Papadokostopoulou A, Mathioudaki K, Scorilas A, et al. Colon cancer and protein arginine methyltransferase 1 gene expression Anticancer Res 2009; 29: 1361-6. |
| [57] | Seligson DB, Horvath S, Shi T, et al. Global histone modification patterns predict risk of prostate cancer recurrence Nature 2005; 435: 1262-6. |
| [58] | Cheung N, Chan LC, Thompson A, Cleary ML, So CWE. Protein arginine-methyltransferase-dependent oncogenesis Nat Cell Biol 2007; 9: 1208-5. |
| [59] | Di Lorenzo A, Bedford MT. Histone arginine methylation FEBS Lett 2010. DOI: 10.1016/j.febslet.2010.11.010 |
| [60] | Majumder S, Liu Y, Ford OH, Mohler JL, Whang YE. Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability Prostate 2006; 66: 1292-301. |
| [61] | Frietze S, Lupien M, Silver PA, Brown M. CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1 Cancer Res 2008; 68: 301-6. |
| [62] | Hong H, Kao CH, Jeng MH, et al. Aberrant expression of CARM1, a transcriptional coactivator of androgen receptor, in the development of prostate carcinoma and androgen-independent status Cancer 2004; 101: 83-9. |
| [63] | Pal S, Vishwanath SN, Erdjument-Bromage H, Tempst P, Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes Mol Cell Biol 2004; 24: 9630-45. |
| [64] | Pal S, Sif S. Interplay between chromatin remodelers and protein arginine methyltransferases J Cell Physiol 2007; 213: 306-15. |
| [65] | Wang L, Pal S, Sif S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells Mol Cell Biol 2008; 28: 6262-77. |
| [66] | Kim JM, Sohn HY, Yoon SY, et al. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells Clin Cancer Res 2005; 11: 473-82. |
| [67] | Verbiest V, Montaudon D, Tautu MT, et al. Protein arginine (N)-methyl transferase 7 (PRMT7) as a potential target for the sensitization of tumor cells to camptothecins FEBS Lett 2008; 582: 1483-9. |
| [68] | Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570. |
| [69] | Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery Nat Rev Drug Discov 2009; 8: 724-32. |
| [70] | Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders Nat Rev Neurosci 2007; 8: 355-67. |
| [71] | Jones PA, Baylin SB. The epigenomics of cancer Cell 2007; 128: 683-92. |
| [72] | Schaefer A, Sampath SC, Intrator A, et al. Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex Neuron 2009; 64: 678-91. |
| [73] | Maze I, Covington HE 3rd, Dietz DM, et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity Science 2010; 327: 213-6. |
| [74] | Imai K, Togami H, Okamoto T. Involvement of histone H3 lysine 9 (H3K9) methyl transferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294 J Biol Chem 2010; 285: 16538-45. |
| [75] | Frye SV. The art of the chemical probe Nat Chem Biol 2010; 6: 159-61. |
| [76] | Zheng YG, Wu J, Chen ZY, Goodman M. Chemical regulation of epigenetic modifications: Opportunities for new cancer therapy Med Res Rev 2008; 28: 645-87. |
| [77] | Mori S, Iwase K, Iwanami N, Tanaka Y, Kagechika H, Hirano T. Development of novel bisubstrate-type inhibitors of histone methyltransferase SET7/9 Bioorg Med Chem 2010; 18: 8158-66. |
| [78] | Osborne T, Roska RLW, Rajski SR, Thompson PR. In situ generation of a bisubstrate analogue for protein arginine methyltransferase 1 J Am Chem Soc 2008; 130: 4574-5. |
| [79] | Dowden J, Hong W, Parry RV, Pike RA, Ward SG. Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases Bioorg Med Chem Lett 2010; 20: 2103-5. |
| [80] | Rea S, Eisenhaber F, O'Carroll D, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases Nature 2000; 406: 593-9. |
| [81] | Min JR, Feng Q, Li ZZ, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase Cell 2003; 112: 711-23. |
| [82] | Frye SV, Heightman T, Jin J. Targeting methyl lysine Annu Rep Med Chem 2010; 45: 329-43. |
| [83] | Qian C, Zhou MM. SET domain protein lysine methyltransferases: Structure, specificity and catalysis Cell Mol Life Sci 2006; 63: 2755-63. |
| [84] | Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7. |
| [85] | Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3. |
| [86] | Fog CK, Jensen KT, Lund AH. Chromatin-modifying proteins in cancer APMIS 2007; 115: 1060-89. |
| [87] | Dambacher S, Hahn M, Schotta G. Epigenetic regulation of development by histone lysine methylation Heredity 2010; 105: 24-37. |
| [88] | Martin C, Zhang Y. The diverse functions of histone lysine methylation Nat Rev Mol Cell Biol 2005; 6: 838-49. |
| [89] | Rathert P, Dhayalan A, Murakami M, et al. Protein lysine methyltransferase G9a acts on non-histone targets Nat Chem Biol 2008; 4: 344-6. |
| [90] | Van Duyne R, Easley R, Wu WL, et al. Lysine methylation of HIV-1 Tat regulates transcriptional activity of the viral LTR Retrovirology 2008; 5: 13. |
| [91] | Pagans S, Kauder SE, Kaehlcke K, et al. The cellular lysine methyltransferase Set7/9-KMT7 binds HIV-1 TAR RNA, monomethylates the viral transactivator Tat, and enhances HIV transcription Cell Host Microbe 2010; 7: 234-44. |
| [92] | Huang J, Dorsey J, Chuikov S, et al. G9a and Glp methylate lysine 373 in the tumor suppressor p53 J Biol Chem 2010; 285: 9636-41. |
| [93] | Kubicek S, O'Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase Mol Cell 2007; 25: 473-81. |
| [94] | Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9 Nat Chem Biol 2005; 1: 143-5. |
| [95] | Iwasa E, Hamashima Y, Fujishiro S, et al. Total synthesis of (+)-chaetocin and its analogues: their histone methyltransferase G9a inhibitory activity J Am Chem Soc 2010; 132: 4078-9. |
| [96] | Gardiner DM, Waring P, Howlett BJ. The epipolythiodioxopiperazine (ETP) class of fungal toxins: distribution, mode of action, functions and biosynthesis Microbiology 2005; 151: 1021-32. |
| [97] | Liu F, Chen X, Allali-Hassani A, et al. Protein lysine methyltransferase G9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines J Med Chem 2010; 53: 5844-7. |
| [98] | Collazo E, Couture JF, Bulfer S, Trievel RC. A coupled fluorescent assay for histone methyltransferases Anal Biochem 2005; 342: 86-92. |
| [99] | Wigle TJ, Provencher LM, Norris JL. Accessing protein methyltransferase and demethylase enzymology using microfluidic capillary electrophoresis Chem Biol 2010; 17: 695-704. |
| [100] | Vedadi M, Barsyte-Lovejoy D, Liu F, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells Nat Chem Biol 2011; 7: 566-74. |
| [101] | For the latest unpublished progress in this area, see: http://www.thesgc.org/chemical_probes/UNC0638/#overview |
| [102] | Chang Y, Ganesh T, Horton JR, et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases J Mol Biol 2010; 400: 1-7. |
| [103] | Lee YH, Stallcup MR. Minireview: Protein arginine methylation of nonhistone proteins in transcriptional regulation Mol Endocrinol 2009; 23: 425-33. |
| [104] | Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S. Protein arginine methyltransferases: Evolution and assessment of their pharmacological and therapeutic potential Pharmacol Ther 2007; 113: 50-87. |
| [105] | Miranda TB, Miranda M, Frankel A, Clarke S. PRMT7 is a member of the protein arginine methyltransferase family with a distinct substrate specificity J Biol Chem 2004; 279: 22902-7. |
| [106] | Bedford MT. Arginine methylation at a glance J Cell Sci 2007; 120: 4243-6. |
| [107] | Lakowski TM, Frankel A. Kinetic analysis of human protein arginine N-methyltransferase 2: formation of monomethyl- and asymmetric dimethyl-arginine residues on histone H4 Biochem J 2009; 421: 253-61. |
| [108] | Cook JR, Lee JH, Yang ZH, et al. FBXO11/PRMT9, a new protein arginine methyltransferase, symmetrically dimethylates arginine residues Biochem Biophys Res Commun 2006; 342: 472-81. |
| [109] | Fielenbach N, Guardavaccaro D, Neubert K, et al. DRE-1: An evolutionarily conserved F box protein that regulates C-elegans developmental age Dev Cell 2007; 12: 443-55. |
| [110] | Pahlich S, Bschir K, Chiavi C, Belyanskaya L, Gehring H. Different methylation characteristics of protein arginine methyltransferase 1 and 3 toward the Ewing sarcoma protein and a peptide Proteins: Struct Funct Bioinform 2005; 61: 164-75. |
| [111] | Cheng DH, Cote J, Shaaban S, Bedford MT. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing Mol Cell 2007; 25: 71-83. |
| [112] | Bedford MT, Reed R, Leder P. WW domain-mediated interactions reveal a spliceosome-associated protein that binds a third class of proline-rich motif: The proline glycine and methionine-rich motif Proc Natl Acad Sci USA 1998; 95: 10602-7. |
| [113] | Bedford MT, Richard S. Arginine methylation: An emerging regulator of protein function Mol Cell 2005; 18: 263-72. |
| [114] | Cheng DH, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases J Biol Chem 2004; 279: 23892-9. |
| [115] | Bonham K, Hemmers S, Lim YH, Hill DM, Finn MG, Mowen KA. Effects of a novel arginine methyltransferase inhibitor on T-helper cell cytokine production FEBS J 2010; 277: 2096-108. |
| [116] | Trapp J, Meier R, Hongwiset D, Kassack MU, Sippl W, Jung M. Structure-activity studies on suramin analogues as inhibitors of NAD(+)-dependent histone deacetylases (sirtuins) ChemMedChem 2007; 2: 1419-31. |
| [117] | Ragno R, Simeoni S, Castellano S, et al. Small molecule inhibitors of histone arginine methyltransferases: Homology modeling, molecular docking, binding mode analysis, and biological evaluations J Med Chem 2007; 50: 1241-53. |
| [118] | Balasubramanyam K, Varier RA, Altaf M, et al. Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription J Biol Chem 2004; 279: 51163-71. |
| [119] | Mai A, Cheng D, Bedford MT, et al. Epigenetic multiple ligands: Mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors J Med Chem 2008; 51: 2279-90. |
| [120] | Mowen KA, Schurter BT, Fathman JW, David M, Glimcher LH. Arginine methylation of NIP45 modulates cytokine gene expression in effector T lymphocytes Mol Cell 2004; 15: 559-71. |
| [121] | Richard S, Morel M, Cleroux P. Arginine methylation regulates IL-2 gene expression: a role for protein arginine methyltransferase 5 (PRMT5) Biochem J 2005; 388: 379-86. |
| [122] | Blanchet F, Cardona A, Letimier FA, Hershfield MS, Acuto O. CD28 costimulatory signal induces protein arginine methylation in T cells J Exp Med 2005; 202: 371-7. |
| [123] | Purandare AV, Chen Z, Huynh T, et al. Pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2008; 18: 4438-1. |
| [124] | Huynh T, Chen Z, Pang S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 2924-7. |
| [125] | Therrien E, Larouche G, Manku S, et al. 1,2-Diamines as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 6725-32. |
| [126] | Allan M, Manku S, Therrien E, et al. N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett 2009; 19: 1218-23. |
| [127] | Wan HH, Huynh T, Pang SH, et al. Benzo[d]imidazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1)-hit to lead studies Bioorg Med Chem Lett 2009; 19: 5063-6. |
| [128] | Spannhoff A, Heinke R, Bauer I, et al. Target-based approach to inhibitors of histone arginine methyltransferases J Med Chem 2007; 50: 2319-5. |
| [129] | Bissinger EM, Heinke R, Sippl W, Jung M. Targeting epigenetic modifiers: Inhibitors of histone methyltransferases Med Chem Commun 2010; 1: 114-24. |
| [130] | Jung M. Epigenetic targets in drug discovery In: Sippl W, Jung M, Eds. Weinheim Wiley-VCH 2009; 251-68. |
| [131] | Heinke R, Spannhoff A, Meier R, et al. Virtual screening and biological characterization of novel histone arginine methyltransferase PRMT1 inhibitors ChemMedChem 2009; 4: 69-77. |
| [132] | Spannhoff A, Machmur R, Heinke R, et al. A novel arginine methyltransferase inhibitor with cellular activity Bioorg Med Chem Lett 2007; 17: 4150-3. |
| [133] | Feng Y, Li MY, Wang BH, Zheng YG. Discovery and mechanistic study of a class of protein arginine methylation inhibitors J Med Chem 2010; 53: 6028-39. |
| [134] | Selvi BR, Batta K, Kishore AH, et al. Identification of a novel inhibitor of coactivator-associated arginine methyltransferase 1 (CARM1)-mediated methylation of histone H3 Arg-17 J Biol Chem 2010; 285: 7143-52. |
| [135] | Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery Nat Rev Drug Discov 2009; 8: 724-32. |
| [136] | Pollock RM, Richon VM. Epigenetic approaches to cancer therapy Drug Discov Today: Ther Strategies 2009; 6: 71-9. |
| [137] | Turanov AA, Lobanov AV, Fomenko DE, et al. Genetic code supports targeted insertion of two amino acids by one codon Science 2009; 323: 259-61. |
| [138] | Kouzarides T, Berger SL. Epigenetics. Cold Spring Harbor: Cold Spring Harbor Laboratory Press 2007; pp. 191-209. |
| [139] | Garcia BA, Hake SB, Diaz RL, et al. Organismal differences in post-translational modifications in histones H3 and H4 J Biol Chem 2007; 282: 7641-55. |
| [140] | Gileadi O, Knapp S, Lee WH, et al. The scientific impact of the Structural Genomics Consortium: a protein family and ligand-centered approach to medically-relevant human proteins J Struct Funct Genomics 2007; 8: 107-9. |
| [141] | Weigelt J, McBroom-Cerajewski LD, Schapira M, Zhao Y, Arrowsmith CH. Structural genomics and drug discovery: all in the family Curr Opin Chem Biol 2008; 12: 32-9. |
| [142] | Hajduk PJ, Greer J. A decade of fragment-based drug design: strategic advances and lessons learned Nat Rev Drug Discov 2007; 6: 211-9. |
| [143] | Everts S. Piece by piece: More and more companies are using fragment-based lead design as a drug discovery strategy Chem Eng News 2008; 86: 15-23. |