- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Development of RNA Aptamer and its Ligand Binding Assay on Microchip Electrophoresis
Ken-ichi Ohnoa, Chikara Nakataa, Yoshihiro Sanoa, Fumiko Nishikawab, Satoshi Nishikawab, Hidetoshi Arakawa*, a
Abstract
Microchip electrophoresis (ME) coupled with fluorescence detection was used to estimate the binding activity of aptamer in each systematic evolution of ligands by exponential enrichment (SELEX) round for a target molecule. This approach is a non-radioisotopic, rapid and simple platform, and electrophoretic separation appears to be an effective technique for aptamers of oligonucleotide molecules. We tried to obtain gonadotropin-specific RNA aptamer by the above approach. As a result, the peaks of aptamers based on the conformational differences between them were separated and detected on the electropherograms. Moreover, the intensity of peak of unbound aptamer was decreased with progression through the SELEX rounds, suggesting that RNA aptamer with high affinity was obtained by the proposed method.
Article Information
Identifiers and Pagination:
Year: 2012Volume: 6
First Page: 1
Last Page: 5
Publisher Id: CCGTM-6-1
DOI: 10.2174/1875397301206010001
Article History:
Received Date: 13/7/2011Revision Received Date: 19/12/2011
Acceptance Date: 23/12/2011
Electronic publication date: 24/1/2012
Collection year: 2012
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the School of Pharmacy, Showa University, 1-5-8 Hatanodai, Shinagawa, Tokyo 142-8555, Japan; Tel: +81-3-3784-8194; Fax: +81-3-3784-8247; E-mail: arakawa@pharm.showa-u.ac.jp
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 13-7-2011 |
Original Manuscript | Development of RNA Aptamer and its Ligand Binding Assay on Microchip Electrophoresis | |
INTRODUCTION
Antigen-antibody reaction is a sensitive and selective biological response against viruses, bacteria and exogenous substances. Antibodies have specific affinity for the corresponding target molecules (antigens), and are considered the most useful and powerful tool for analytical, biochemical and clinical techniques, such as enzyme-linked and radioimmunoassay, immunoprecipitation, immunostaining and western blotting, because their target molecules are easily identified and captured from complex matrices, including blood, cells, tissues, and other biological and artificial fluids. In recent years, several biosensors have been developed that use antibody coupled with the detection systems of surface plasmon resonance (SPR) [1 Soelberg SD, Stevens RC, Limaye AP, Furlong CE. Surface plasmon resonance detection using antibody-linked magnetic nanoparticles for analyte capture, purification, concentration, and signal amplification Anal Chem 2009; 81: 2357-63.], quartz crystal microbalance (QCM) [2 Shen Z, Mernaugh RL, Yan H, Yu L, Zhang Y, Zeng X. Engineered recombinant single-chain fragment variable antibody for immunosensors Anal Chem 2005; 77: 6834-42.] and fluorescence resonance energy transfer (FRET) [3 Enomoto K, Araki A, Nakajima T, et al. High-throughput miniaturized immunoassay for human interleukin-13 secreted from NK3.3 cells using homogenous time-resolved fluorescence J Pharm Biomed Anal 2002; 28: 73-9.]. However, it is difficult and requires a lot of effort and time to prepare and obtain target-specific antibodies, especially monoclonal antibodies, because of in vivo materials from animal sources, in addition, the difficulty depends on the target molecules and the biological properties of immunological tolerance and toxic potential. These constraints place limits on the range of antibodies available to us.
On the other hand, aptamers are single-stranded DNA and RNA oligonucleotides that fold into unique secondary or ternary structures, such as hairpin, bulge, pseudoknot and G-quartet, which have the potential to capture a ligand (target) molecule similarly to antibodies. By in vitro selection, namely, systematic evolution of ligands by exponential enrichment (SELEX) [4 Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment : RNA ligands to Bacteriophage T4 DNA polymerase Science 1990; 249: 505-10., 5 Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands Nature 1990; 346: 818-22.], it is possible in principle to prepare and obtain aptamers easily for unlimited target molecules. There have been several different kinds of aptamers developed for amino acids, medicines, nucleic acids, vitamins, peptides and proteins [6 Stoltenburg R, Reinemann C, Strehlitz B. SELEX-a (r)evolutionary method to generate high-affinity nucleic acid ligands Biomol Eng 2007; 24: 381-403.]. The SELEX approach is based on affinity selection from a very large DNA or RNA library with random sequences. In general, SELEX is carried out by the following three-step process that forms a single round, which consists of (i) the isolation of DNAs or RNAs bound to target molecules from a reaction mixture of DNA or RNA library by affinity-based techniques, (ii) the amplification of the isolated DNAs or RNAs by (RT-)PCR and (iii) the estimation of the affinity for a target molecule. Several assay techniques such as nitrocellulose filtration, affinity chromatography and affinity bead-based assay have been employed for studies on the binding activities of aptamers with their target molecules, but these techniques only enable semi-quantitative measurement.
In our previous report, aptamer-based binding assays with intercalating dyes of ethidium bromide (EtBr), SYBR Gold and SYBR Green II have already been described using microchip electrophoresis (ME) coupled with fluorescence detection [7 Nishikawa F, Arakawa H, Nishikawa S. Application of microchip electrophoresis in the analysis of RNA aptamer-protein interactions Nucleosides Nucleotides Nucleic Acids 2006; 25: 369-82.]. ME and capillary electrophoresis are useful for many kinds of ligand binding assays because of their high separation efficiency as a B/F separation system and the minimal sample volume. Our approach has the advantages that the unbound (free) aptamer can be separated from aptamer-protein complex on a microchip in a short time, and there is no need to prepare modifications such as fluorescence labeling and immobilization as shown in Fig. (1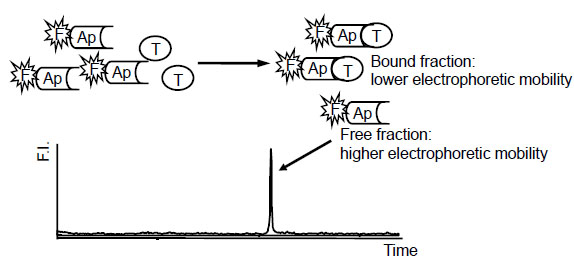 ). It was observed that the intensity of peaks of unbound aptamers was decreased with the increase in the corresponding protein concentration, and then each calibration curve was obtained with high sensitivity as expected. In this study, by using ME to estimate the binding activity in SELEX rounds, we have tried to develop an efficient novel RNA aptamer for human menopausal gonadotropin (hMG) related to human chorionic gonadotropin (hCG) and applied it to aptamer-based binding assay, since our previous report demonstrates the clinical significance of developing a sensitive immunoassay of hCG for use in quality control and diagnosis [8 Arakawa H, Shiokawa M, Imamura O, Maeda M. Novel bioluminescent assay of alkaline phosphatase using adenosine-3’-phosphate-5’phosphosulfate as substrate and the luciferin-luciferase reaction and its application Anal Biochem 2003; 314: 206-11.].
). It was observed that the intensity of peaks of unbound aptamers was decreased with the increase in the corresponding protein concentration, and then each calibration curve was obtained with high sensitivity as expected. In this study, by using ME to estimate the binding activity in SELEX rounds, we have tried to develop an efficient novel RNA aptamer for human menopausal gonadotropin (hMG) related to human chorionic gonadotropin (hCG) and applied it to aptamer-based binding assay, since our previous report demonstrates the clinical significance of developing a sensitive immunoassay of hCG for use in quality control and diagnosis [8 Arakawa H, Shiokawa M, Imamura O, Maeda M. Novel bioluminescent assay of alkaline phosphatase using adenosine-3’-phosphate-5’phosphosulfate as substrate and the luciferin-luciferase reaction and its application Anal Biochem 2003; 314: 206-11.].
 |
Fig. (1) Principle of aptamer-based binding assay by microchip electrophoresis: Ap, aptamer; F, fluorescent intercalating dye; T, target molecule; F.I., fluorescence intensity. |
EXPERIMENTAL
Materials and Methods
Template DNA for RNA library and primers for PCR amplification and RNA synthesis were obtained from Takara Bio Inc. (Shiga, Japan). Template DNA (60 bases) was designed with primer sequences of 15 bases at both ends for PCR amplification and a randomized sequence of 30 bases in the middle region. The sequences are as follows:
template DNA: 5’-GGTAGATACGATGGA-N30-CATGAC GCGCAGCCA-3’, 5’-end primer: 5’-AGTAATACGACTC ACTATAGGTAGATACGATGGA-3’ (the underlined sequence is T7 promoter region), 3’-end primer: 5’-TGGCTGCGCGTCATG-3’.
SELEX Procedure
SELEX was carried out in accordance with a previous report. In brief, the single-stranded template DNA (4.5 nmol) was converted into double-stranded DNA by PCR, by the following procedure: 94 °C for 1 min (94 °C for 1 min, 55 °C for 1 min, 72 °C for 1 min) x 3 cycles, with 3’-end primer (4.5 nmol). The reaction mixture was re-amplified by PCR, by the following procedure: (94 °C for 1 min, 55 °C for 1 min, 72 °C for 1 min) x 3 cycles, with 5’-end and 3’-end primers (1 nmol each), and the resultant solution was purified by ethanol precipitation. For RNA synthesis, PCR product of 79 base pairs was subjected to in vitro transcription with T7 RNA polymerase (T7 Ampliscribe kit, Epicentre Technology) and purified by spin down with a Bio-spin 30 (Bio-Rad Laboratories, Hercules, CA). The obtained 60 bases RNA library was used for SELEX, in which the appropriate amount of RNA was dissolved with 50 mmol/L Tris-HCl buffer (pH 7.6) containing 5 mmol/L MgCl2, 25 mmol/L NaCl and 25 mmol/L KCl, and was heated at 90 °C for 2 min and then slowly cooled to room temperature. The RNA solution was premixed with target molecule (hMG) and incubated at room temperature for 0.5–60 min, and the reaction mixture was passed through a nitrocellulose filter (HAWP filter, 0.45 μm, Millipore). The RNAs bound to hMG were retained on the filter, and recovered with the same buffer heated at 90 °C for 5 min. The recovered RNA was again passed through a fresh nitrocellulose filter to remove proteins and RNAs nonspecifically bound to the filter. The eluted RNA was recovered and purified by ethanol precipitation. The purified RNA was reverse-transcribed with avian myeloblastosis virus (AMV, Wako Pure Chemicals) with 3’-end primer at 42 °C for 60 min, and the obtained cDNA was amplified by PCR, 94 °C for 1 min (94 °C for 1 min, 55 °C for 0.75 min, 72 °C for 1 min) x 10–15 cycles, with 5’-end and 3’-end primers. The PCR product was recovered by ethanol precipitation, and transcribed into RNA with T7 RNA polymerase at 37 °C for 2 h, and the reaction mixture was purified by 8% (w/v) polyacrylamide gel electrophoresis (PAGE) on gel containing 7 mol/L urea. The amount of newly obtained RNA library was determined by absorption spectrometry at 260 nm and subjected to the next SELEX round.
Microchip Electrophoresis
For aptamer-based binding assay with RNA obtained in each round, ME was performed using a cosmo-i SV1100 from Hitachi Electronics Engineering Co. Ltd. (Tokyo, Japan), and a Hitachi Chemical microchip, which is designed in a cross type channels for the injection with a 10 mm and the separation with a 45 mm in length, a 0.1 mm in width and a 0.03 mm in depth, and is fabricated with polymethyl methacrylate (PMMA) as shown in Fig. (2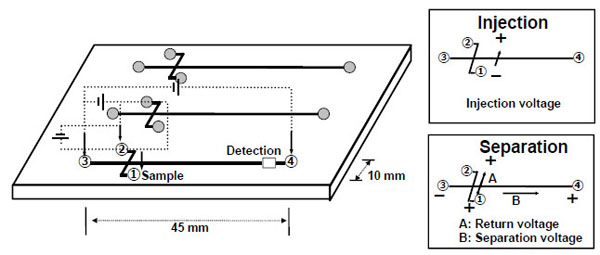 ). The channel was filled with 0.5% hydroxypropyl methylcellulose (HPMC, Sigma-Aldrich, St. Louis, MO) in 1 x TBE buffer containing 0.5 µg/mL EtBr or 10,000-fold dilution of SYBR Green II (Invitrogen, Carlsbad, CA) for fluorescence detection. The sample solutions were prepared with RNA libraries and hMG protein as described in the SELEX procedure and were incubated for 30 min at room temperature; then, fluorescein as the internal standard was added before the measurements. The sample solution (10 μL) was subjected to ME in triplicate.
). The channel was filled with 0.5% hydroxypropyl methylcellulose (HPMC, Sigma-Aldrich, St. Louis, MO) in 1 x TBE buffer containing 0.5 µg/mL EtBr or 10,000-fold dilution of SYBR Green II (Invitrogen, Carlsbad, CA) for fluorescence detection. The sample solutions were prepared with RNA libraries and hMG protein as described in the SELEX procedure and were incubated for 30 min at room temperature; then, fluorescein as the internal standard was added before the measurements. The sample solution (10 μL) was subjected to ME in triplicate.
 |
Fig. (2) Schematic illustration of microchip electrophoresis. |
The electrophoresis was performed with an injection voltage of 300 V for 60 sec, and at separation and return voltages of 750 V and 180 V, respectively, for 180 sec simultaneously. On the obtained electropherograms, the peak height for RNA was normalized with the peak height for fluorescein as the internal standard.
RESULTS AND DISCUSSION
RNA template (60 bases) was initially obtained from single-stranded DNA template (60 bases) by PCR and subsequent in vitro transcription, and was subjected to 12 successive rounds of selection. In the first round of selection, the randomized RNA library, which contains approximately 1015 molecules (4.5 nmol), was mixed and incubated with 0.5 nmol hMG. As SELEX rounds progressed, the binding conditions were restricted to refine RNA library to those RNAs with higher affinity for hMG. Over the whole SELEX rounds, the reaction time was gradually decreased; in addition, the amounts of template RNAs and hMG and number of PCR cycles were decreased in the last two rounds. As a result, specific RNA libraries were obtained in each round, and the detailed SELEX conditions and the resultant amounts of recovered RNA are shown in Table 1.
Next, to estimate the binding activity of each round of RNAs, the reaction mixture of RNAs and hMG was subjected to ME using EtBr for ligand binding assay after each round of selection. Typical electropherograms are shown in Fig. (3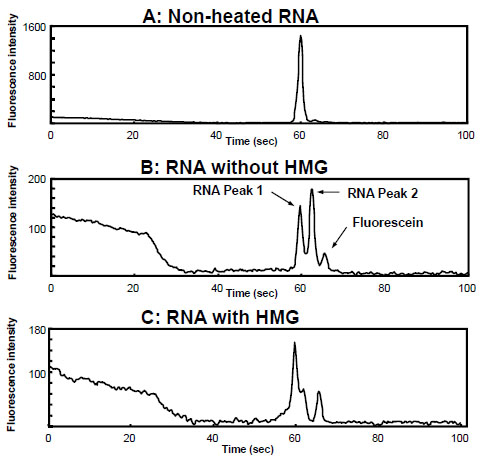 ), which were produced using the 11th generation of RNA (G11 RNA). As shown in Fig. (3A), an electropherogram of one peak around 60 sec was obtained using unheated G11 RNA alone. However, the heated G11 RNA provided two major peaks (Fig. 3B), suggesting that, by heating and slowly cooling, RNA molecules would fold into unique secondary and ternary conformations (functional structures) from a non-specific single-stranded conformation. This result is consistent with our previous reports of the separation and detection of single nucleotide polymorphisms (SNPs) by capillary electrophoresis on the basis of single-strand DNA conformation polymorphism (SSCP) [9 Arakawa H, Nakashiro S, Maeda M, Tsuji A. Analysis of single-strand DNA conformation polymorphism by capillary electrophoresis J Chromatogr A 1996; 722: 359-68., 10 Arakawa H, Igarashi H, Kashiwazaki H, Maeda M, Tokita A, Yamashiro Y. Single-stranded conformation polymorphism analysis of Vitamin D recepter gene by capillary electrophoresis with laser-induced fluorescence detection Anal Chim Acta 2001; 445: 197-204.]. Furthermore, the different kinds of conformations could be efficiently separated on ME. Fig. (3C) shows the result of the reaction mixture of G11 RNA and hMG, indicating that the second peak (peak 2) of G11 RNA was decreased in the presence of hMG, but the first peak (peak 1) showed little change. Therefore, peak 2 should correspond to the RNAs with higher affinity for hMG compared with peak 1. On the other hand, no peak corresponding to RNA-hMG complex was observed under the proposed separation mode and conditions. Fig. (4
), which were produced using the 11th generation of RNA (G11 RNA). As shown in Fig. (3A), an electropherogram of one peak around 60 sec was obtained using unheated G11 RNA alone. However, the heated G11 RNA provided two major peaks (Fig. 3B), suggesting that, by heating and slowly cooling, RNA molecules would fold into unique secondary and ternary conformations (functional structures) from a non-specific single-stranded conformation. This result is consistent with our previous reports of the separation and detection of single nucleotide polymorphisms (SNPs) by capillary electrophoresis on the basis of single-strand DNA conformation polymorphism (SSCP) [9 Arakawa H, Nakashiro S, Maeda M, Tsuji A. Analysis of single-strand DNA conformation polymorphism by capillary electrophoresis J Chromatogr A 1996; 722: 359-68., 10 Arakawa H, Igarashi H, Kashiwazaki H, Maeda M, Tokita A, Yamashiro Y. Single-stranded conformation polymorphism analysis of Vitamin D recepter gene by capillary electrophoresis with laser-induced fluorescence detection Anal Chim Acta 2001; 445: 197-204.]. Furthermore, the different kinds of conformations could be efficiently separated on ME. Fig. (3C) shows the result of the reaction mixture of G11 RNA and hMG, indicating that the second peak (peak 2) of G11 RNA was decreased in the presence of hMG, but the first peak (peak 1) showed little change. Therefore, peak 2 should correspond to the RNAs with higher affinity for hMG compared with peak 1. On the other hand, no peak corresponding to RNA-hMG complex was observed under the proposed separation mode and conditions. Fig. (4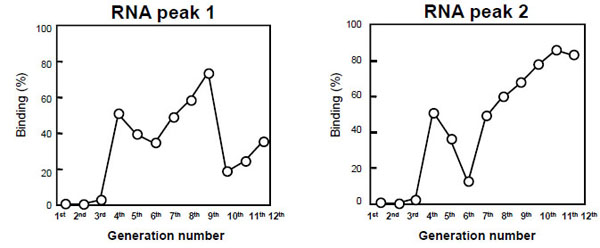 ) shows the binding activities of G1–G12 RNAs obtained from the peak heights of peaks 1 and 2. The binding activities of peak 2 were improved with the increase in the rounds of selection. Among G1–G12 RNAs, peak 2 of G11 RNA showed the highest binding activity for hMG, so G11 RNA was applied to aptamer-based binding assay with EtBr and SYBR Green II for highly sensitive fluorescence detection. As a result, the intensity of peak 2 of G11 RNA was decreased with an increase in hMG concentration, and then the standard curves for hMG were obtained over the concentration range of 1–200 pmol/assay using EtBr, and 0.1–10 pmol/assay using SYBR Green II as shown in Fig. (5
) shows the binding activities of G1–G12 RNAs obtained from the peak heights of peaks 1 and 2. The binding activities of peak 2 were improved with the increase in the rounds of selection. Among G1–G12 RNAs, peak 2 of G11 RNA showed the highest binding activity for hMG, so G11 RNA was applied to aptamer-based binding assay with EtBr and SYBR Green II for highly sensitive fluorescence detection. As a result, the intensity of peak 2 of G11 RNA was decreased with an increase in hMG concentration, and then the standard curves for hMG were obtained over the concentration range of 1–200 pmol/assay using EtBr, and 0.1–10 pmol/assay using SYBR Green II as shown in Fig. (5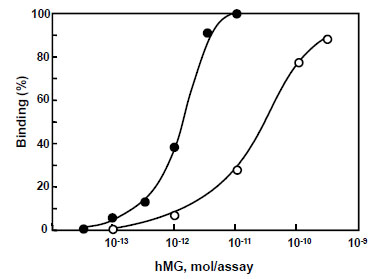 ). The reasons for the sensitivity are that SYBR Green II exhibits higher fluorescence quantum yield with single-stranded RNA, and its excitation wavelength is suitable for the blue LED of the light source on the device. In the proposed aptamer-based binding assays, 50 pmol/assay RNA was used with SYBR Green II compared with the assay with EtBr in which 200 pmol/assay RNA was used. Finally, the proposed assay was applied to hMG in pharmaceutical preparations for injection. Since protein (hormonal) activities alone were found in their package insert irrespective of the hMG concentration level, their dilution curves were evaluated. As a result, linear curves were obtained without matrix effects from two samples (data not shown). It was demonstrated that G11 RNA could exhibit a level of performance in the binding assay equal to that of antibody.
). The reasons for the sensitivity are that SYBR Green II exhibits higher fluorescence quantum yield with single-stranded RNA, and its excitation wavelength is suitable for the blue LED of the light source on the device. In the proposed aptamer-based binding assays, 50 pmol/assay RNA was used with SYBR Green II compared with the assay with EtBr in which 200 pmol/assay RNA was used. Finally, the proposed assay was applied to hMG in pharmaceutical preparations for injection. Since protein (hormonal) activities alone were found in their package insert irrespective of the hMG concentration level, their dilution curves were evaluated. As a result, linear curves were obtained without matrix effects from two samples (data not shown). It was demonstrated that G11 RNA could exhibit a level of performance in the binding assay equal to that of antibody.
 |
Fig. (3) Electropherograms for aptamer-based binding assays. |
 |
Fig. (4) Binding activities of aptamers obtained in each round for hMG. |
 |
Fig. (5) Standard curves for hMG with EtBr (open circle) and SYBR Green II (closed circle). |
In conclusion, by the 12 successive rounds of selection and the binding assay on ME, G11 RNA was obtained as an hMG-specific aptamer. ME is a rapid and simple platform for a binding assay compared with conventional assays such as filter binding assay and affinity chromatography. The electrophoretic separation appears to be an effective technique for oligonucleotide molecules, and it could be a useful approach for efficient aptamer development to fractionate DNA and RNA molecules on the basis of conformational differences. We have been conducting further studies on the potential use of capillary electrophoresis and ME for aptamer development.
CONFLICTS OF INTEREST
None Declared.
ACKNOWLEDGEMENT
None Declared.