- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
A Dual Reporter Splicing Assay Using HaloTag-containing Proteins
Koichi Oshimaa, b, g, Takahiro Nagasea, Kohsuke Imaic, Shigeaki Nonoyamac, Megumi Obarad, Tomoyuki Mizukamid, Hiroyuki Nunoid, Hirokazu Kaneganee, Futoshi Kuribayashif, Shin Amemiyag, Osamu Ohara*, a, b
Abstract
To evaluate the effects of genetic variations on mRNA splicing, we developed a minigene-based splicing assay using reporter genes encoding luciferase and the multifunctional HaloTag protein. In addition to conventional RT-PCR analysis, splicing events can be monitored in this system using two parameters: luciferase activity and signals derived from HaloTag-containing proteins bound to a fluorescent ligand following SDS-PAGE. The luciferase activity reflects the accumulated amounts of successfully spliced HaloTag–luciferase fusion products, whereas the amounts and sizes of HaloTag-containing proteins provide quantitative insights into precursor, correctly spliced, and aberrantly spliced mRNA species. Preliminary experiments confirmed that the dual reporter minigene assay can provide estimates of overall splicing efficiency based on the levels of protein products. We then used the minigene assay to analyze a case of chronic granulomatous disease that was caused by a G>C mutation at position +5 in the 5’-splice donor site of intron 5 of the CYBB gene. We found that the G>C mutation affected CYBB mRNA splicing by changing a delicate balance of splicing efficiencies of introns 4, 5, and 6.
Article Information
Identifiers and Pagination:
Year: 2012Volume: 6
Issue: Suppl 1
First Page: 27
Last Page: 37
Publisher Id: CCGTM-6-27
DOI: 10.2174/1875397301206010027
Article History:
Received Date: 02/3/2012Revision Received Date: 09/5/2012
Acceptance Date: 16/5/2012
Electronic publication date: 20/9/2012
Collection year: 2012
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Department of Human Genome Research, Kazusa DNA Research Institute, Kisarazu, Chiba Japan; Tel: +81-438-52-3913; Fax: +81-483-52-3914; E-mail: ohara@kazusa.or.jp
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 02-3-2012 |
Original Manuscript | A Dual Reporter Splicing Assay Using HaloTag-containing Proteins | |
INTRODUCTION
Determining whether mutations at splice sites cause a disease generally involves comparative reverse transcription-PCR (RT-PCR) analysis of the patient and the patient’s family members using appropriate RNA samples. The necessary RNA samples, however, are not always available in practice. Thus, alternative approaches with surrogate minigene constructs have been widely used to test the effects of mutations on splicing [1Cooper TA. Use of minigene systems to dissect alternative splicing elements Methods 2005; 37: 331-40.-3Andreassi C, Jarecki J, Zhou J, et al. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients Hum Mol Genet 2001; 10: 2841-9.]. Whereas conventional (or end-point) RT-PCR is the method of choice to monitor splicing events in most minigene-based approaches, this technique does not allow robust quantitative analysis and routine estimates of overall splicing efficiency. Although quantitative RT-PCR may overcome some of these limitations, designing a quantitative RT-PCR system for precursor, correctly spliced, and aberrantly spliced products without a priori knowledge of the splicing events is extremely challenging.
Reporter-based minigene assays can also be used to quantitatively evaluate the effects of genetic variation on splicing because the reporter gene allows monitoring of splicing events based on the levels of protein products. Previously described reporter-based minigene systems, however, can detect only normally spliced products and not aberrant products generated because of cryptic splice-site activation, exon skipping, or intron retention, for example. To overcome this problem, we developed a dual reporter system that includes luciferase and the new tag protein HaloTag (molecular mass, approximately 33 kDa; [4Los GV, Wood K. The HaloTag: a novel technology for cell imaging and protein analysis Methods Mol Biol 2007; 356: 195-208.]). Luciferase activity can be used as a quantitative measure of correctly spliced minigene products, whereas the HaloTag moiety, which can be easily detected just by adding a fluorescent HaloTag ligand to cell lysates prior to SDS-PAGE, allows us to quantify unspliced, correctly spliced, and aberrantly spliced minigene products without protein blotting [4Los GV, Wood K. The HaloTag: a novel technology for cell imaging and protein analysis Methods Mol Biol 2007; 356: 195-208.].
MATERIALS AND METHODOLOGY
Characterization of Abnormal Phenotypes in a Patient with Chronic Granulomatous Disease (CGD)
All experiments with human samples were reviewed and approved by the appropriate Institutional Review Boards. Production of reactive oxygen species in neutrophils obtained from a patient with CGD was analyzed using flow cytometry, the fluorescent probe dihydrorhodamine 123 (DHR-123), and phorbol myristate acetate as the stimulus. Neutrophils were also examined for surface expression of cytochrome b558 using 7D5 anti-cytochrome b558 monoclonal antibodies and phycoerythrin-conjugated secondary antibodies. The stained neutrophils were analyzed using FACScan and CellQuest software (Becton Dickinson, San Jose, CA). We also analyzed the CYBB gene of the patient after obtaining informed consent. Coding exons and flanking intronic sequences were amplified in PCRs using a Multiplex PCR Assay Kit (Takara Bio Inc., Shiga, Japan) (Supplementary Table 1). PCR products were purified using an ExoSAP-IT kit (GE Healthcare, Amersham, UK), and directly sequenced using an ABI 3130 DNA sequencer (Life Technologies, Carlsbad, CA). cDNA was created using neutrophils from the patient, his mother, and a healthy donor, and subjected to RT-PCR analysis of a region spanning exons 4 to 8 (upstream primer: CGAAGACAACTGGACAGGAA-TC; downstream primer: GTGATGACCACCTTCTGTTGAGAT). We directly sequenced amplified products excised from agarose gels.
Construction of Exon/Intron Cassette-trapping Plasmids Carrying the HaloTag and Renilla Luciferase Genes
We first amplified the coding region of the Renilla luciferase gene from pRL-CMV (Promega, Madison, WI) using a high-fidelity polymerase (KOD plus ver. 2 PCR kit; Toyobo, Osaka, Japan). The PCR product was cloned into the SgfI and PmeI sites of pFN21A to create pFN21A-RL (Promega). The exon/intron cassette of interest was prepared using KOD plus ver. 2 PCR kit (Toyobo) with 20-bp flanking extensions that were homologous to the vector ends, based on instructions from an In-Fusion kit (Clontech Laboratories, Mountain View, CA). The cassette was designed to terminate translation in an intronic region when the cassette was inserted between the HaloTag and luciferase genes, whereas in-frame splicing at authentic splicing sites in the cassette generated HaloTag–luciferase fusion proteins. The exon/intron cassette was purified on an agarose gel and cloned into the SgfI site of pFN21A-RL using an In-Fusion protocol (Clontech Laboratories). Detailed experimental conditions are described in Supplementary Table
2 and the overall scheme is shown in Fig. (1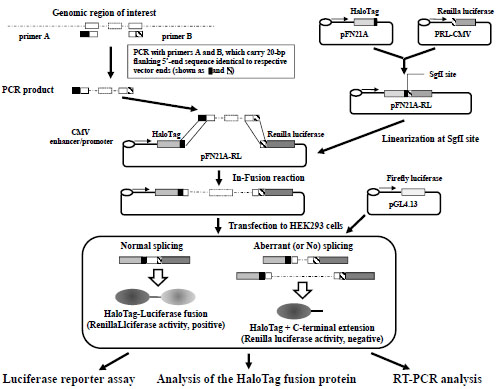 ).
).

Wild-type exon/intron cassettes were prepared in PCRs using commercially available human genomic DNA from a healthy donor (Clontech Laboratories) as a template. Unless stated otherwise, mutant exon/intron cassettes were prepared from patient genomic DNA (Supplementary Table 2). In some cases, specific nucleotide substitutions were introduced using PCR-assisted overlap-extension mutagenesis [5Ho SN, Hunt HD, Horton RM, et al. Site-directed mutagenesis by overlap extension using the polymerase chain reaction Gene 1989; 77: 51-9.]. The resulting reporter plasmids were subjected to DNA sequenceing using a BigDye Terminator Ver. 3 Kit (Life Technologies) to confirm the sequences of the inserted fragments.
Transfection
Unless stated otherwise, the splicing assay was conducted using human embryonic kidney 293T cells that were seeded in 100 µ l of growth medium (Dulbecco’s modified Eagle medium–high glucose supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin-amphotericin B solution; Gibco Invitrogen, Carlsbad, CA, USA) at 2 × 104 cells/well in 96-well plates. Twenty-four hours after plating, cells were transfected with 100 ng of reporter plasmid and 2 ng of pGL4.13 encoding the firefly luciferase reporter (Promega) using FuGENE HD (Roche Diagnostics, Indianapolis, IN, USA) according to the manufacturer’s recommendations. After 16–24 h, HaloTag fusion proteins were labeled with 5 µM HaloTag tetramethylrhodamine (TMR) ligand (Promega) and incubated for an additional 20–24 h in a 5% CO2 incubator at 37°C. The percentage of TMR-positive cells was determined using a Biozero fluorescence microscope (Keyence, Osaka, Japan) with a TRITC filter setting (OP-66837, Keyence).
Luciferase Reporter Assay
Dual luciferase assays were performed using the Dual-Luciferase Reporter Assay System (Promega) 44 h following transfection. After mixing 10 µl of cell extracts with 100 µl of Luciferase Assay Reagent II in a 96-well plate, firefly luciferase activity was measured using a GloMax 96 Microplate Luminometer (Promega). Following the first measurement, Renilla luciferase activities were measured with the same luminometer after 100 µl of Stop and Glo reagent was added (Promega). All samples were assayed in triplicate. Average Renilla luciferase activities for the wild-type and mutant exon/intron cassettes in the same experimental trial were compared after normalizing the samples based on firefly luciferase activities to compensate for variations in transfection efficiency.
Analysis of HaloTag Fusion Proteins
Extracts from TMR ligand–treated cells prepared as described for the luciferase reporter assay were separated on a 12% polyacrylamide gel. TMR fluorescent signals were detected and quantified using a FLA-3000 system (Fujifilm, Tokyo, Japan) and MultiGauge image analysis software (Fujifilm).
RT-PCR Analysis
cDNA was synthesized using the ImProm II Reverse Transcription System (Promega) and total RNA (approximately 200 ng) that was prepared with an RNeasy Plus Micro Kit (Qiagen, Valencia, CA) 48 h after cells were transfected. The resulting cDNA samples served as templates in 20-μl PCR reactions using a Multiplex PCR Assay Kit (Takara Bio, Kyoto, Japan) according to the manufacturer’s instructions. The upstream and downstream PCR primers and RT-PCR conditions are described in Supplementary Table 3. PCR products were separated and quantified using a MCE-202 MultiNA Microchip Electrophoresis System (Shimadzu Biotech, Kyoto, Japan).
Statistical Analysis
Data are expressed as means ± standard errors. Unless stated otherwise, statistical significance was determined using Student's t-tests and defined as P values < 0.05.
RESULTS
1. Development of a Dual Reporter–based Splicing Assay to Evaluate the Effects of Splice-site Mutations
a. A Dual Reporter–based Splicing Assay
Fig. (1) illustrates the overall scheme that was used to construct a dual reporter minigene plasmid. Each exon/intron cassette was amplified from genomic DNA, and inserted into a dual reporter minigene vector. The resultant plasmids were introduced into cells together with a plasmid encoding firefly luciferase to normalize transfection efficiencies in each experiment. HaloTag proteins were monitored using a membrane-permeable fluorescent HaloTag ligand (TMR-HaloTag ligand). Because binding between the TMR-HaloTag ligand and the HaloTag moiety is highly stoichiometric (1:1 ratio), the fluorescence intensities of protein bands after SDS-PAGE likely reflected relative levels of HaloTag proteins after cell lysis. Thus, luciferase activity was proportional to the amount of properly spliced HaloTag–luciferase fusion proteins, whereas unspliced and aberrantly spliced products were quantified based on fluorescent signals from protein bands after SDS-PAGE and labeling of HaloTag-containing proteins. Conventional RT-PCR analysis can also be performed with this system; however, although RT-PCR analysis is useful for characterizing spliced transcripts, quantification of transcripts using conventional RT-PCR assays is limited because of differences in PCR amplification efficiencies for various mRNA species. In particular, because unspliced mRNA is frequently large enough to considerably lower amplification efficiencies, end-point RT-PCR assays are often used to estimate the relative amounts of alternatively spliced mRNA. Although quantitative RT-PCR assays may provide a solution to this problem, this technique is somewhat laborious owing to the need to prepare control DNA to quantify all spliced and unspliced products.
b. Evaluation of a G>T Mutation at Position –1 of the 3’-splicing Acceptor Site of Intron 16 in the Gene Encoding Bruton’s Tyrosine Kinase (BTK)
As a proof-of-concept experiment, we first examined the effects of a G>T mutation at position –1 of the 3’-splicing acceptor site of intron 16 of the BTK gene, which was identified as a causative mutation for X-linked agammaglobulinemia based on a genetic test performed at Kazusa DNA Research Institute. This mutation disrupts the canonical GT-AG rule of splicing boundaries and, as expected, resulted in complete loss of mature RT-PCR products owing to a lack of splicing of intron 16 (Fig. 2 ). Consistent with this result, we observed almost complete suppression of luciferase activity and very low levels of the HaloTag–luciferase fusion protein with the mutant minigene construct. The lower fluorescent band shown in Fig. (2) was likely a product of minigene precursor mRNA, because the molecular mass estimated based on its electrophoretic mobility was consistent with the deduced molecular mass of the translation product of the precursor mRNA (i.e., a HaloTag protein with a carboxy-terminal extension of 16 amino-acid residues). The splicing efficiency of this BTK exon/intron cassette in the minigene construct was estimated from the relative fluorescent HaloTag band intensities with and without luciferase. Although end-point RT-PCR can also be used to estimate splicing efficiency, this method can produce biased results due to differences in amplification efficiencies. Because measuring fluorescence intensities of HaloTag bands is simple and direct, the dual reporter assay appears to provide more consistent analysis of overall splicing efficiency compared with end-point RT-PCR assays.
). Consistent with this result, we observed almost complete suppression of luciferase activity and very low levels of the HaloTag–luciferase fusion protein with the mutant minigene construct. The lower fluorescent band shown in Fig. (2) was likely a product of minigene precursor mRNA, because the molecular mass estimated based on its electrophoretic mobility was consistent with the deduced molecular mass of the translation product of the precursor mRNA (i.e., a HaloTag protein with a carboxy-terminal extension of 16 amino-acid residues). The splicing efficiency of this BTK exon/intron cassette in the minigene construct was estimated from the relative fluorescent HaloTag band intensities with and without luciferase. Although end-point RT-PCR can also be used to estimate splicing efficiency, this method can produce biased results due to differences in amplification efficiencies. Because measuring fluorescence intensities of HaloTag bands is simple and direct, the dual reporter assay appears to provide more consistent analysis of overall splicing efficiency compared with end-point RT-PCR assays.

c. Evaluation of Mutations at Position +5 of the 5’-splicing Donor Site of Intron 6 in the Gene Encoding Wiskott-Aldrich Syndrome Protein (WAS)
We next examined the effects of nucleotide changes (G>A, G>T, or G>C) at position +5 of the 5’-splicing donor site of intron 6 in the WAS gene. Although only the G>A change has been described in case reports [6Falet H, Hoffmeister KM, Neujahr R, Hartwig JH. Normal Arp2/3 complex activation in platelets lacking WASp Blood 2002; 100: 2113-2.-14Zhu Q, Watanabe C, Liu T, et al. Wiskott-Aldrich syndrome/X-linked thrombocytopenia: WASP gene mutations, protein expression, and phenotype Blood 1997; 90: 2680-9.], we examined the effects of all possible nucleotide changes at this position. Fluorescence images of polyacrylamide gels indicated that all mutant minigene constructs generated a band in addition to those derived from mature and precursor mRNA (Fig. 3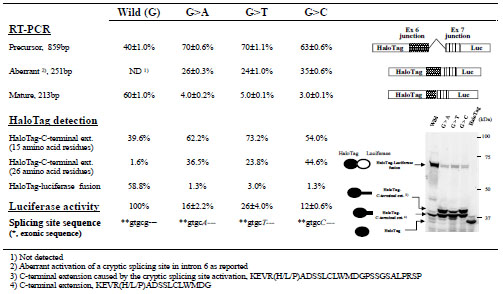 ). Judging from the size of the additional band, it did not include a luciferase moiety and was likely an aberrantly spliced product. Sequencing the RT-PCR products revealed that all of the mutations activated a cryptic splicing donor site in the dual reporter splicing assay, as has been observed in vivo [6Falet H, Hoffmeister KM, Neujahr R, Hartwig JH. Normal Arp2/3 complex activation in platelets lacking WASp Blood 2002; 100: 2113-2.-14Zhu Q, Watanabe C, Liu T, et al. Wiskott-Aldrich syndrome/X-linked thrombocytopenia: WASP gene mutations, protein expression, and phenotype Blood 1997; 90: 2680-9.]. The nucleotide sequence of the RT-PCR products suggested that the protein in the additional band included a carboxy-terminal extension of 26 amino-acid residues. This result indicated that SDS-PAGE analysis can be used to identify aberrant splicing without DNA sequencing. Furthermore, our dual reporter assay revealed that, the cryptic site was used even with the wild-type minigene construct, whereas this splicing event was not detected in a conventional RT-PCR assay. Moreover, the protein products revealed that mutation of this site increased not only the amounts of aberrantly spliced mRNA at the cryptic splicing site but also those of precursor mRNA. Finally, although no marked differences in the effects of the G>A, G>T, and G>C mutations were detected using a conventional RT-PCR assay, our dual reporter assay showed that the G>C mutation produced the greatest effects on normal splicing, whereas the G>T mutation produced the smallest effects.
). Judging from the size of the additional band, it did not include a luciferase moiety and was likely an aberrantly spliced product. Sequencing the RT-PCR products revealed that all of the mutations activated a cryptic splicing donor site in the dual reporter splicing assay, as has been observed in vivo [6Falet H, Hoffmeister KM, Neujahr R, Hartwig JH. Normal Arp2/3 complex activation in platelets lacking WASp Blood 2002; 100: 2113-2.-14Zhu Q, Watanabe C, Liu T, et al. Wiskott-Aldrich syndrome/X-linked thrombocytopenia: WASP gene mutations, protein expression, and phenotype Blood 1997; 90: 2680-9.]. The nucleotide sequence of the RT-PCR products suggested that the protein in the additional band included a carboxy-terminal extension of 26 amino-acid residues. This result indicated that SDS-PAGE analysis can be used to identify aberrant splicing without DNA sequencing. Furthermore, our dual reporter assay revealed that, the cryptic site was used even with the wild-type minigene construct, whereas this splicing event was not detected in a conventional RT-PCR assay. Moreover, the protein products revealed that mutation of this site increased not only the amounts of aberrantly spliced mRNA at the cryptic splicing site but also those of precursor mRNA. Finally, although no marked differences in the effects of the G>A, G>T, and G>C mutations were detected using a conventional RT-PCR assay, our dual reporter assay showed that the G>C mutation produced the greatest effects on normal splicing, whereas the G>T mutation produced the smallest effects.

2. Using Dual Reporter Splicing Assays to Characterize an Intronic Mutation in the CYBB Gene of a Patient with CGD
a. Identification of a Mutation in a Patient with CGD
The patient was a 2-year-old boy with no family history of CGD who had been hospitalized several times because of Bacillus Calmette-Guérin vaccine–induced regional axillary lymphadenitis, liver abscesses, and bacterial lymphadenitis. Flow cytometry with DHR-123 showed reduced reactive oxygen species production in activated neutrophils from the patient (Fig. 4A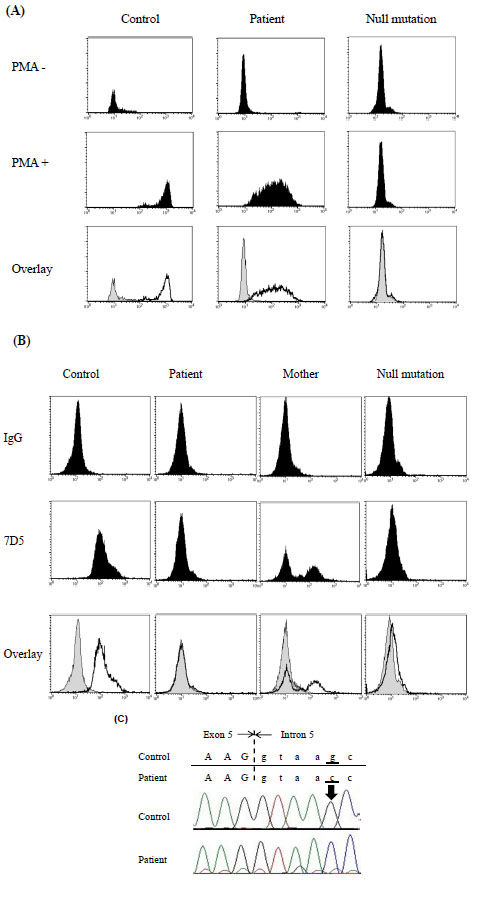 ). On the other hand, the patient’s neutrophils showed markedly decreased expression of cytochrome b558 and the mother’s neutrophils exhibited a mosaic pattern (Fig. 4B); because of random X chromosome inactivation, female carriers of genes that cause X-linked CGD often exhibit this type of cytometric pattern. After obtaining informed consent, protein coding exons and short flanking intronic regions of the CYBB gene were directly sequenced. We identified a single G>C nucleotide substitution at position +5 in the 5’-splice donor site of intron 5 (Fig. 4C), a site at which a single nucleotide G>A substitution was previously identified [15Rae J, Newburger PE, Dinauer MC, et al. X-Linked chronic granulomatous disease: mutations in the CYBB gene encoding the gp91-phox component of respiratory-burst oxidase Am J Hum Genet 1998; 62: 1320-31.].
). On the other hand, the patient’s neutrophils showed markedly decreased expression of cytochrome b558 and the mother’s neutrophils exhibited a mosaic pattern (Fig. 4B); because of random X chromosome inactivation, female carriers of genes that cause X-linked CGD often exhibit this type of cytometric pattern. After obtaining informed consent, protein coding exons and short flanking intronic regions of the CYBB gene were directly sequenced. We identified a single G>C nucleotide substitution at position +5 in the 5’-splice donor site of intron 5 (Fig. 4C), a site at which a single nucleotide G>A substitution was previously identified [15Rae J, Newburger PE, Dinauer MC, et al. X-Linked chronic granulomatous disease: mutations in the CYBB gene encoding the gp91-phox component of respiratory-burst oxidase Am J Hum Genet 1998; 62: 1320-31.].

We then characterized the effects of this newly identified mutation in the CYBB gene using clinical samples. We performed RT-PCRs to amplify exon 4 to exon 8 in cDNA created from the neutrophils of the patient, his mother, and a healthy donor (Fig. 5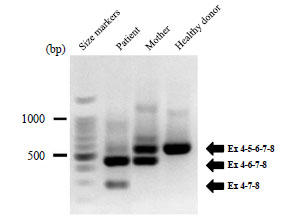 ). The RT-PCR products were separated on an agarose gel and directly sequenced, revealing exon skipping of CYBB exon 5 or exons 5 and 6 in the patient, whereas only heterozygous skipping of exon 5 was observed in his mother. These results indicated that the mutation caused CGD due to exon skipping. We then evaluated the effects of the mutation on CYBB splicing events quantitatively using the new dual reporter minigene assay.
). The RT-PCR products were separated on an agarose gel and directly sequenced, revealing exon skipping of CYBB exon 5 or exons 5 and 6 in the patient, whereas only heterozygous skipping of exon 5 was observed in his mother. These results indicated that the mutation caused CGD due to exon skipping. We then evaluated the effects of the mutation on CYBB splicing events quantitatively using the new dual reporter minigene assay.

b. Evaluating Mutations at Position +5 of the 5’-splicing Donor Site of Intron 5 in the CYBB Gene Using Dual Reporter Minigene Assays
We used our splicing assay to examine splicing of CYBB intron 5 flanked by short regions of exons 5 and 6 with and without a nucleotide change at position +5 of the 5’-splicing donor site (exon/intron cassettes containing a single intron). Although these experimental conditions were similar to those used in the preliminary experiment with the WAS gene (Fig. 3), RT-PCR and luciferase assays indicated that changing the nucleotide from G to another residue did not lower the splicing efficiency. Interestingly, the absolute luciferase activity observed with the minigene construct containing the G>C change was reproducibly highest among the minigene constructs (Fig. 6 ). In addition, the splicing efficiency observed with this cassette appeared high because the amount of HaloTag fusion protein without luciferase (< 5% of total protein; Fig. 6), which was likely transcribed from precursor mRNA, were lower than results observed with cassettes containing the BTK or WAS gene (24.2% or 39.6% of total protein, respectively). HaloTag fusion proteins without luciferase appeared as a doublet on polyacrylamide gels even though we expected that a single product of approximately that size was encoded by the minigene construct. The mutant cassettes resulted in slight but detectable increases in the amount of luciferase-deficient HaloTag products (Fig. 6), suggesting a small decrease in splicing efficiency with the exon/intron cassette carrying a mutation at the +5 position of CYBB intron 5. Thus, the mutation found in the patient with CGD did not markedly affect the splicing of intron 5.
). In addition, the splicing efficiency observed with this cassette appeared high because the amount of HaloTag fusion protein without luciferase (< 5% of total protein; Fig. 6), which was likely transcribed from precursor mRNA, were lower than results observed with cassettes containing the BTK or WAS gene (24.2% or 39.6% of total protein, respectively). HaloTag fusion proteins without luciferase appeared as a doublet on polyacrylamide gels even though we expected that a single product of approximately that size was encoded by the minigene construct. The mutant cassettes resulted in slight but detectable increases in the amount of luciferase-deficient HaloTag products (Fig. 6), suggesting a small decrease in splicing efficiency with the exon/intron cassette carrying a mutation at the +5 position of CYBB intron 5. Thus, the mutation found in the patient with CGD did not markedly affect the splicing of intron 5.

c. Examining the Effects of Mutations at Position +5 of the 5’-splicing Donor Site of CYBB Intron 5 Using Dual Reporter Minigene Constructs with Multiple Introns
Evaluating transcripts from the patient’s neutrophils suggested that the G>C substitution resulted in skipping of exon 5 (major) or exons 5 and 6 (minor; Fig. 5). To reproduce exon skipping using the minigene construct, we next examined two additional exon/intron cassettes: the first cassette contained intron 4–exon 5–intron 5 flanked by short regions of exons 4 and 6 (3950 bp), whereas the second cassette carried intron 4–exon 5–intron 5–exon 6–intron 6 flanked by short regions of exons 4 and 7 (6940 bp). We examined four cassettes containing introns 4 and 5, each with a different nucleotide (G, C, T, or A) at position +5 of the 5’-splicing donor site of CYBB intron 5. On the other hand, only the G>C mutation was examined in the exon/intron cassettes containing three introns.
For mutant exon/intron cassettes containing two or three introns, most RT-PCR products showed the expected exon skipping, whereas both wild-type cassettes also generated a considerable amount of RT-PCR products that were shortened by exon skipping (Figs. 7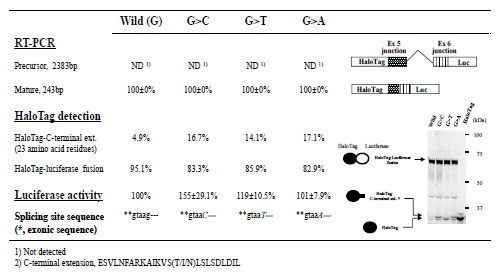 and 8
and 8 ). The results were not consistent with the RT-PCR analyses of blood samples from the patient and family members, which did not show exon skipping for the wild-type CYBB gene, whereas the mutant CYBB allele generated aberrant mRNA without exon 5 (major) or exons 5 and 6 (minor). Analyzing luciferase activities obtained with the exon/intron cassettes containing multiple introns demonstrated that amounts of mature mRNA encoding HaloTag(luciferase fusions decreased by an order of magnitude when one additional intron was included. Thus, SDS-PAGE analysis of HaloTag fusion proteins was of little use in this case because the main protein products produced from the exon/intron cassettes were encoded by partially spliced and/or precursor mRNA. On the other hand, RT-PCR assays allowed us to analyze only spliced mRNA because the introns were too large to be effectively amplified, resulting in a rough estimate of relative amounts of spliced variants derived from each exon/intron cassette. Therefore, only the luciferase assay measured the overall splicing efficiency of each exon/intron cassette (Figs. 7 and 8). The results indicated that the G>C mutation at position +5 of the 5’-splicing donor site of intron 5 markedly reduced the amounts of successfully spliced products relative to those translated after exon skipping, whereas notable amounts of luciferase activities were detected with the mutant exon/intron cassettes (Figs. 7 and 8). It should be also noted that skipping exon 5 was rare in the minigene construct shown in Fig. (8), whereas it was more common than skipping exons 5 and 6 in the patient sample (Fig. 5).
). The results were not consistent with the RT-PCR analyses of blood samples from the patient and family members, which did not show exon skipping for the wild-type CYBB gene, whereas the mutant CYBB allele generated aberrant mRNA without exon 5 (major) or exons 5 and 6 (minor). Analyzing luciferase activities obtained with the exon/intron cassettes containing multiple introns demonstrated that amounts of mature mRNA encoding HaloTag(luciferase fusions decreased by an order of magnitude when one additional intron was included. Thus, SDS-PAGE analysis of HaloTag fusion proteins was of little use in this case because the main protein products produced from the exon/intron cassettes were encoded by partially spliced and/or precursor mRNA. On the other hand, RT-PCR assays allowed us to analyze only spliced mRNA because the introns were too large to be effectively amplified, resulting in a rough estimate of relative amounts of spliced variants derived from each exon/intron cassette. Therefore, only the luciferase assay measured the overall splicing efficiency of each exon/intron cassette (Figs. 7 and 8). The results indicated that the G>C mutation at position +5 of the 5’-splicing donor site of intron 5 markedly reduced the amounts of successfully spliced products relative to those translated after exon skipping, whereas notable amounts of luciferase activities were detected with the mutant exon/intron cassettes (Figs. 7 and 8). It should be also noted that skipping exon 5 was rare in the minigene construct shown in Fig. (8), whereas it was more common than skipping exons 5 and 6 in the patient sample (Fig. 5).


DISCUSSION
Minigene-based splicing assays should quantitatively examine unspliced, aberrantly spliced, and correctly spliced products to determine relative splicing efficiencies with and without mutations. We have developed a dual reporter assay that uses luciferase and HaloTag proteins. Because splicing determines the sequence and length of any carboxy-terminal extension on the HaloTag protein, separating HaloTag fusion proteins encoded by unspliced, aberrantly spliced, and correctly spliced mRNA via SDS-PAGE allowed us to estimate the amounts of various protein products by measuring the fluorescent intensities of the protein bands. Although the relationships between electrophoretic mobilities of proteins during SDS-PAGE and the proteins’ molecular masses are complex, the degree of protein migration usually allowed us to determine which band contained each of the possible minigene products. In particular, protein products translated from correctly spliced mRNA were easily identified based on the 36-kDa Renilla luciferase moiety at the carboxy terminus, which could be independently evaluated using luciferase activity.
The advantage of the HaloTag reporter was demonstrated using two examples (Figs. 2 and 3). Spicing events were best detailed by integrating data from RT-PCRs, luciferase assays, and fluorescence gel imaging of HaloTag proteins. In each of the preliminary experiments, the data indicated that a single base substitution resulted in a substantial reduction in successful splicing. Of note, spicing efficiencies (i.e., the percentages of spliced products) estimated using RT-PCRs and HaloTag-based SDS-PAGE assays were similar, whereas the splicing efficiency observed with the BTK exon/intron cassette was considerably different from that detected with the WAS exon/intron cassette; although mutation of the canonical GT-AG splicing junction in the BTK gene almost completely disrupted splicing (Fig. 2), mutation at position +5 of the 5’-splicing donor site of the WAS gene activated a cryptic splicing site in the intron and lowered the splicing efficiency from 60% to 30–40% based on RT-PCR and SDS-PAGE analyses (Fig. 3). For larger introns, however, precursor mRNA may not be amplified in RT-PCRs, as in the case of the CYBB gene (Figs. 6–8). Instead, the HaloTag moiety and a fluorescently labeled ligand can be used to monitor overall splicing efficiencies without biases caused by differences in the lengths of intronic sequences. This is the most prominent advantage of the HaloTag moiety as a reporter in the minigene-based splicing assay.
In the last experiment, we characterized the effects of a newly identified mutation in the CYBB gene of a patient with CGD using our minigene-based splicing assay. When we examined the effects of this mutation using an exon/intron cassette containing only intron 5, no inhibition of splicing was observed. The data shown in Fig. (6) indicated that splicing efficiency observed with this exon/intron cassette was highest among the examined cassettes (approximately 95%) and the mutation at position +5 of the splicing donor site slightly lowered the splicing efficiency. On the contrary, luciferase activity levels showed slight increases in the amounts of successfully spliced HaloTag–luciferase fusion proteins (Fig. 6), suggesting that overall expression of HaloTag–luciferase fusion proteins was enhanced by the mutation. The results indicated that the mutation did not markedly affect splicing efficiency of intron 5. We, however, observed exon skipping in CYBB transcripts from the patient. Thus, using the minigene-based assay and cassettes with a single intron to monitor spicing events based on the “intron definition model” [16Hertel KJ. Combinatorial control of exon recognition J Biol Chem 2008; 283: 1211-5.] was inappropriate for this case. Instead, we examined the effects of the G>C mutation on splicing using exon/intron cassettes carrying two and three introns (Figs. 7 and 8, respectively). Significant exon skipping was observed even with the wild-type cassette, suggesting that inclusion of exon 5 in transcripts is intrinsically opportunistic; the results shown in Figs (7 and 8) indicated that inclusion of exon 5 seemed to be governed by a delicate balance of splicing efficacies of flanking introns. This splicing profile cannot be explained based solely on the local genomic context of the splicing site. For example, although mutations at position +5 of the 5'-splicing donor site have frequently been reported to produce a range of effects in various genes [17Buratti E, Chivers M, Kralovicova J, et al. Aberrant 5' splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization Nucleic Acids Res 2007; 35: 4250-63.-19Kralovicova J, Vorechovsky I. Global control of aberrant splice-site activation by auxiliary splicing sequences: evidence for a gradient in exon and intron definition Nucleic Acids Res 2007; 35: 6399-413.], the identified mutation in this case was not the clear cause of the splicing abnormalities based on an evaluation with genomic analysis software (MutationTaster; www.mutation-taster.org/), which makes predictions based on local sequences around the splicing sites.
Other data obtained using the minigene assay were also somewhat discrepant from the patient data; in the minigene system, skipping exons 5 and 6 was more prominent than skipping exon 5 or exon 6 (Fig. 8). These results likely reflected, at least in part, lower rates of splicing for each intron when the transcripts were transiently expressed in the minigene system compared with in vivo processes. Thus, the levels of spliced products in the minigene assay depend not only on splicing efficiencies of various introns but also on the number of splicing events required to generate the products; mRNA that is derived from the cassettes and lacks exon 5 or 6 is generated through two splicing steps, whereas a single splicing event can produce mRNA without exons 5 and 6 (Fig. 8). In addition, nonsense-mediated mRNA decay can affect steady-state levels of aberrantly spliced products in vivo. Because total HaloTag-specific signals were almost constant regardless of changes in splicing patterns, nonsense-mediated mRNA decay likely does not play a prominent role in our minigene assay. The results obtained using the minigene assay, however, were qualitatively consistent with those observed in vivo. In fact, the Human-transcriptome Database for Alternative Splicing (www.h-invitational.jp/h-dbas/) shows that exons 2, 5, and 6 of the CYBB gene are sometimes skipped, even in wild-type transcripts.
CONFLICT OF INTEREST DISCLOSURE
The authors have no financial conflicts of interest regarding the material discussed in the manuscript.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article Download Supplementary Material.
ACKNOWLEDGEMENTS
This work was supported by a Grant-in-Aid for Scientific Research; the program for Fostering Regional Innovation from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; and a grant for research on intractable diseases from the Ministry of Health, Labour, and Welfare of Japan. We also thank the Kazusa DNA Research Institute (Chiba, Japan) and the RIKEN Research Center for Allergy and Immunology (Yokohama, Japan) for continued support.
REFERENCES
Endorsements
Browse Contents
Table of Contents
- INTRODUCTION
- MATERIALS AND METHODOLOGY
- RESULTS
- Development of a Dual Reporter–based Splicing Assay to Evaluate the Effects of Splice-site Mutations
- DISCUSSION
- CONFLICT OF INTEREST DISCLOSURE