- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Open Chemistry Journal
(Discontinued)
ISSN: 1874-8422 ― Volume 8, 2021
Synthesis, Characterization and Antifungal Assessment of Optically Active Bis-organotin Compounds Derived from (S)-BINOL Diesters
Andrea R. Costantino1, Jörg M. Neudörfl2, Romina A. Ocampo1, Laura A. Svetaz3, Susana A. Zacchino3, Liliana C. Koll1, Sandra D. Mandolesi1, *
Abstract
Background:
Organotin(IV) derivatives have appeared recently as potential biologically active metallopharmaceuticals exhibiting a variety of therapeutic activities. Hence, it is important to study the synthesis of new organotin compounds with low toxicity that may be of pharmacological interest.
Objectives:
This study focuses on the synthesis of new bis-stannylated derivatives with C2 symmetry that could be tested as antifungal agents against two clinical important fungal species, Cryptococcus neoformans and Candida albicans.
Methods:
The radical addition of triorganotin hydrides (R3SnH) and diorganotin chlorohydrides (R2ClSnH) to bis-α,β-unsaturated diesters derived from (S)-BINOL led to the corresponding new bis-stannylated derivatives with C2 symmetry. Nine pure organotin compounds were synthesized with defined stereochemistry. Four of them were enantiomerically pure and four were diastereoisomeric mixtures.
Results:
All new organotin compounds were fully characterized, those with phenyl ligands bonded to tin were the most active compounds against both the strains (Cryptococcus neoformans and Candida albicans), with activity parameters of IC50 close to those of the reference drug (amphotericin B).
Conclusion:
Nine pure organotin compounds with C2 symmetry were synthesized with defined stereochemistry and their antifungal properties were tested against two clinical important fungi with IC values close to those of the reference drug. The structure-containing preferably two or three phenyl groups joined to the tin atom were highly active against both the strains compared with those possessing tri-n-butyl groups.
Article Information

Identifiers and Pagination:
Year: 2019Volume: 6
First Page: 34
Last Page: 46
Publisher Id: CHEM-6-34
DOI: 10.2174/1874842201906010034
Article History:
Received Date: 20/02/2019Revision Received Date: 05/04/2019
Acceptance Date: 22/04/2019
Electronic publication date: 31/05/2019
Collection year: 2019
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 International Public License (CC-BY 4.0), a copy of which is available at: https://creativecommons.org/licenses/by/4.0/legalcode. This license permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
* Address correspondence to this author at the Department of INQUISUR, Departamento de Química, Universidad Nacional del Sur (UNS)- CONICET, Av. Alem 1253, 8000 Bahía Blanca, Argentina; Tel: +54-291-459-5101;
Fax: +54-291-459-5187; Email: sdmando@criba.edu.ar
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 20-02-2019 |
Original Manuscript | Synthesis, Characterization and Antifungal Assessment of Optically Active Bis-organotin Compounds Derived from (S)-BINOL Diesters | |
1. INTRODUCTION
Organotin compounds have been used for many years in different applications, for example as catalysts, heat and light stabilizers, additives of PVC, antifouling, antimold and germicidal agents [1Hoch, M. Organotin compounds in the environment - An overview. Appl. Geochem., 2001, 16, 719-743.
[http://dx.doi.org/10.1016/S0883-2927(00)00067-6] -5Davies, A.G. Tin Chemistry: Fundamentals, Frontiers and Applications, 2008, ]. After near 170 years of their discovery, Tin(IV) organic compounds are one of the most important concerning organometallic chemistry researches [6Smith, P.J., Ed.; Chemistry of Tin., (2nd ed. ), 1997, -10Takahashi, H.; Yasui, S.; Tsunoi, S.; Shibata, I. Catalytic cycloaddition of 2-methyleneaziridines with 1,1-dicyanoalkenes. Org. Lett., 2014, 16(4), 1192-1195.
[http://dx.doi.org/10.1021/ol500062a] [PMID: 24495160] ]. Their toxicity depends on the alkyl group, decreasing with the increase in the alkyl chain length [11Thoonen, S.H.L.; Deelman, B-J.; van Koten, G. Synthetic aspects of tetraorganotins and organotin(IV) halides. J. Organomet. Chem., 2004, 689, 2145-2157.
[http://dx.doi.org/10.1016/j.jorganchem.2004.03.027] ]. In the last fifteen years, organotin(IV) derivatives have appeared also as potential biologically active metallopharmaceuticals exhibiting antitumor, antibacterial, anti-inflammatory and antimicrobial activity [12Hadjikakou, S.K.; Hadjiliadis, N. Antiproliferative and anti-tumor activity of organotin compounds. Coord. Chem. Rev., 2009, 253, 235-249.
[http://dx.doi.org/10.1016/j.ccr.2007.12.026] , 13Iqbal, H.; Ali, S.; Shahzadi, S. Antituberculosis study of organotin(IV) complexes: A review. Cogent Chem., 2015, 1, 1029039.
[http://dx.doi.org/10.1080/23312009.2015.1029039] ]. Considering the limitations caused by its toxicity, it is important to develop new organotin compounds that could be safer and more effective for chemotherapeutic uses.
Taking into account that bis-chlorostannanes Ph2ClSn– R–SnClPh2 show interesting biological activities [14Thodupunoori, S.K.; Alamudun, I.A.; Cervantes-Lee, F.; Gomez, F.D.; Carrasco, Y.P.; Pannell, K.H. Synthesis, structures and preliminary biological screening of bis(diphenyl)chlorotin complexes and adducts: Ph2ClSn-CH 2-R-CH2-SnClPh2, R = p-C6H4, CH2CH2. J. Organomet. Chem., 2006, 69, 11790-11796.
[http://dx.doi.org/10.1016/j.jorganchem.2005.12.040] ], we present here the synthesis and antifungal assessment of optically active organotin compounds derived from (S)-BINOL as part of our research work on the behavior of C2 symmetry diol diesters in radical hydrostannations [15Costantino, A.R.; Ocampo, R.A.; Montiel Schneider, M.G.; Fernandez, G.; Koll, L.C.; Mandolesi, S.D. Efficient routes to racemic and enantiomerically pure (S)-BINOL diesters. Synth. Commun., 2013, 43, 3192-3202.
[http://dx.doi.org/10.1080/00397911.2013.774017] ]. The aim of these studies was to determine the scope and limitations of the change in the structural core of the chiral diol over the stereoselectivity in the radical tandem cyclohydrostannation, which lead to eleven-membered macrodiolides [16Gerbino, D.C.; Koll, L.C.; Mandolesi, S.D.; Podestá, J.C. Stereoselective radical tandem cyclo hydrostannation of optically active di-unsaturated esters of TADDOL. Organometallics, 2008, 27, 660-665.
[http://dx.doi.org/10.1021/om700693p] -18Zacconi, F.C.; Ocampo, R.A.; Podestá, J.C.; Koll, L.C. Synthesis of organotin substituted tricyclic macrodiolides. J. Braz. Chem. Soc., 2016, 27, 484-492.]. The starting materials for building these cycles are open chain systems with two carbon-carbon double bonds activated for radical addition by electron withdrawing substituents like the ester group, as shown in Fig. (1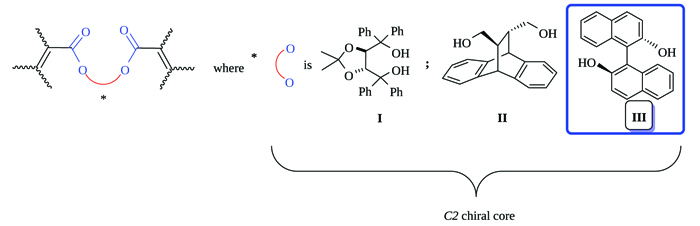 ). The chiral cores employed in these studies were TADDOL (I) and (11R,12R)-9,10-dihydro- 9,10-ethaneanthracene-11,12-dimethanol (II), both C2 symmetry diols. Now, in the present study, we added (S)-BINOL (III).
). The chiral cores employed in these studies were TADDOL (I) and (11R,12R)-9,10-dihydro- 9,10-ethaneanthracene-11,12-dimethanol (II), both C2 symmetry diols. Now, in the present study, we added (S)-BINOL (III).
1,1’-Bi-2-naphthol (BINOL) is an axially chiral organic compound that can be found as a racemic mixture or in its two atropoisomeric forms: M or (R) and P or (S) (Fig. 2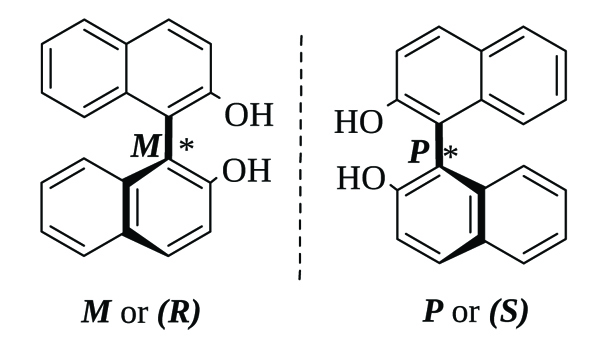 ). Since the chirality axis is not affected in the esterification reactions, the (S)-BINOL derivatives retain such configuration and optical purity.
). Since the chirality axis is not affected in the esterification reactions, the (S)-BINOL derivatives retain such configuration and optical purity.
The internal relative spatial arrangement of bioactive molecules is crucial regarding their ability to interact favorably with active and allosteric sites. Although since 1990, single enantiomers have clearly dominated over racemates in the Drug Discovery and Regulatory Agency approval trends [19Laplante, S.R.; D Fader, L.; Fandrick, K.R.; Fandrick, D.R.; Hucke, O.; Kemper, R.; Miller, S.P.; Edwards, P.J. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem., 2011, 54(20), 7005-7022.
[http://dx.doi.org/10.1021/jm200584g] [PMID: 21848318] ], it is surprising that biologically active atropoisomers have not been extensively covered in the literature, despite their prevalence and importance to the pharmaceutical industry. There are several well-documented atropoisomeric compounds with demonstrated utility as effective drugs. Among them, it is worth mentioning that the glycopeptide P-isomer vancomycin is often used as a drug of penicillin-resistant bacterial infections. It is produced by the soil bacteria Amycolatopsis orientalis, and exhibits broad-spectrum activity against a number of gram-positive bacteria [20McHenry, M.C.; Gavan, T.L. Vancomycin. Pediatr. Clin. North Am., 1983, 30(1), 31-47.
[http://dx.doi.org/10.1016/S0031-3955(16)34318-8] [PMID: 6338468] ]. Another example is gossypol that is produced by Gossypium hirsutum, which has been shown to have possible applications as an antifertility and anticancer agent through binding to antiapoptotic protein Bcl-X L [21Keshmiri-Neghab, H.; Goliaei, B. Therapeutic potential of gossypol: an overview. Pharm. Biol., 2014, 52(1), 124-128.
[http://dx.doi.org/10.3109/13880209.2013.832776] [PMID: 24073600] ]. It has recently been shown that the active atropoisomer of gossypol is that of the M-(-)-enantiomer [22Sprogøe, K.; Staek, D.; Ziegler, H.L.; Jensen, T.H.; Holm-Møller, S.B.; Jaroszewski, J.W. Combining HPLC-PDA-MS-SPE-NMR with circular dichroism for complete natural product characterization in crude extracts: levorotatory gossypol in Thespesia danis. J. Nat. Prod., 2008, 71(4), 516-519.
[http://dx.doi.org/10.1021/np800010r] [PMID: 18290629] ]. As an antecedent of antifungal activity, the atropoisomeric flavans myristinins, isolated from Myristica cinnamomea fruits, showed antifungal activity against Candida albicans with IC50 values ranging from 5.9 to 8.8 µg/mL [23Sawadjoon, S.; Kittakoop, P.; Kirtikara, K.; Vichai, V.; Tanticharoen, M.; Thebtaranonth, Y. Atropisomeric myristinins: selective COX-2 inhibitors and antifungal agents from Myristica cinnamomea. J. Org. Chem., 2002, 67(16), 5470-5475.
[http://dx.doi.org/10.1021/jo020045d] [PMID: 12153244] ].
In the last decades, fungi have emerged as a major cause of human morbidity and mortality, mainly among the immunocompromised and seriously ill-hospitalized patients [24Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev., 2007, 20(1), 133-163.
[http://dx.doi.org/10.1128/CMR.00029-06] [PMID: 17223626] ]. Most of the mycoses-related deaths are associated with the spp. C. albicans and Cryptococcus neoformans and although there are several antifungal drugs in clinical use to fight against this opportunistic fungal spp, the fungal infections remain very difficult to eradicate. C. albicans is among the most common cause of opportunistic fungal infections in immunocompromised hosts. C. neoformans is the most frequent cause of meningitis and is one of the most important HIV-related fatal opportunistic mycoses, which has killed more than 650,000 immunocompromised patients worldwide up to date [25Butts, A.; Krysan, D.J. Antifungal drug discovery: something old and something new. PLoS Pathog., 2012, 8(9)e1002870
[http://dx.doi.org/10.1371/journal.ppat.1002870] [PMID: 22969422] ]. Besides, C. neoformans remains an important life-threatening complication, particularly for immunocompromised patients who have undergone transplantation of solid organs and therefore, new compounds acting against this fungus are highly welcome [26Gutch, R.S.; Nawange, S.R.; Singh, S.M.; Yadu, R.; Tiwari, A.; Gumasta, R.; Kavishwar, A. Antifungal susceptibility of clinical and environmental Cryptococcus neoformans and Cryptococcus gattii isolates in Jabalpur, a city of Madhya Pradesh in Central India. Braz. J. Microbiol., 2015, 46(4), 1125-1133.
[http://dx.doi.org/10.1590/S1517-838246420140564] [PMID: 26691471] ]. Although the incidence of the disease tends to decline in countries with highly active antiretroviral therapy, the outcome of infection is influenced by a variety of factors including the antifungal resistance and new strategies including new structural types with anti-cryptococcal activity are highly welcome [27Trpković, A.; Pekmezović, M.; Barać, A.; Crnčević Radović, L.; Arsić Arsenijević, V. In vitro antifungal activities of amphotericin B, 5-fluorocytosine, fluconazole and itraconazole against Cryptococcus neoformans isolated from cerebrospinal fluid and blood from patients in Serbia. J. Mycol. Med., 2012, 22(3), 243-248.
[http://dx.doi.org/10.1016/j.mycmed.2012.06.002] [PMID: 23518082] ].
In this work, the synthesized atropoisomers (S)-3, (S)-4, (S)-8, (S)-9 and the diastereomeric mixtures 5a-d, 6a-d, 10a-d, 11a-d were tested as antifungal agents against the two mentioned clinical important fungal species, C. neoformans and C. albicans.
 |
Fig. (1) System of two activated carbon-carbon double bonds linked by a C2 chiral core. |
 |
Fig. (2) M or (R) and P or (S) BINOL atropoisomers. |
2. MATERIALS AND METHODS
Unless otherwise noted, all the reagents were purchased in analytical reagent grade from commercial suppliers and used without purification. All the reactions were carried out under inert atmosphere. The solvents used were distilled and dried in accordance with standard procedures. Di-n-butyl- and diphenyltin dihydride were obtained by the reduction of the corresponding dichloride with lithium aluminum hydride [28Kerk, G.J.M.; Noltes, J.G.; Luijten, J.G.A. Investigations on organotin compounds. VIII. Preparation of some organotin hydrides. J. Appl. Chem. (Lond.), 1957, 7, 366-369.
[http://dx.doi.org/10.1002/jctb.5010070704] ] and the starting BINOL unsaturated diesters were prepared as described previously [15Costantino, A.R.; Ocampo, R.A.; Montiel Schneider, M.G.; Fernandez, G.; Koll, L.C.; Mandolesi, S.D. Efficient routes to racemic and enantiomerically pure (S)-BINOL diesters. Synth. Commun., 2013, 43, 3192-3202.
[http://dx.doi.org/10.1080/00397911.2013.774017] ]. Thin layer chromatography was performed on Merck silica gel 60 F254 plates and visualization was accomplished with UV light and/or 5% ethanol solution of phosphomolybdic acid. Silica gel (Merck, 230–400 mesh) was used for column chromatography. Melting points were recorded on a Büchi Melting Point B-545 instrument and are uncorrected. NMR spectra were recorded on a Bruker Avance 300 Multinuclear instrument, using CDCl3 as a solvent; chemical shifts (δ) were reported in ppm with respect to TMS in 1HNMR and 13CNMR, and with respect to Me4Sn in the case of 119SnNMR spectra. Data were reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quin = quintuplet, sex = sextet and br = broad), coupling constant (J values in Hz) and integration.Chemical shifts (δ) are reported in ppm and coupling constants (J) are in Hz. Infrared spectra were recorded with a Nicolet Nexus 470 FT spectrometer. Optical rotations were measured on a Polar L-P, IBZ Messtechnik polarimeter at 589 nm. Compounds described here were characterized by 1H, 13C and DEPT-NMR. Elemental analyses (C, H) were performed in an Exeter analytical, Model CE440 Analyzer.
2.1. General Procedure for the Addition of Triorganotin Hydrides to BINOL Unsaturated Diesters Initiated by AIBN at 75°C
Diester (1 mmol), triorganotin hydride (2.4 mmol) and a catalytic amount of AIBN were stirred in dry toluene (24 mL) at 75°C for 1 hour. The reaction was monitored by TLC and IR spectroscopy. The solvent was then distilled off under reduced pressure. The crude product was thus obtained directly purified by column chromatography using silica gel.
2.1.1. (S)-1,1'-binaphthalene-2,2'-diyl-bis[3-(tri-n-buthylestannyl)propanoate] (S)-3
The product (S)-3 was eluted with hexane/ethyl acetate (98:2) as a yellow oil in a 67% yield. [α]D25 -11.33 (c = 0.45, CHCl3). 1HNMR (300 MHz,CDCl3) δ: 0.71-0.88 (m, 6 x CH2, 6 x CH3, 30H), 0.86-1.00 (m, 6 x CH2, 12H), 1.11-1.31 (m, 6 x CH2, 12H), 1.40 (t, 2J(H, Sn)= 60.57, Hz, 3J(H,H)= 8.71, 2 x CH2, 4H), 2.46 (t, 3J(H, Sn)= 11.8, Hz, 3J(H,H)= 8.71, 2 x CH2, 4H), 7.03-7.12 (m, Ar-H, 4H), 7.15-7.37 (m, Ar-H, 6H), 7.77-7.99 (m, Ar-H, 2H). 13CNMR (75.4 MHz, CDCl3) δ: 2.72 (468.8), 8.83 (323.2), 13.80, 27.39 (56.3), 29.14 (19.5), 31.17 (16.4), 110.82, 117.76, 124.04, 124.20, 127.48, 128.41, 129.45, 131.43, 133.39, 151.74, 173.62 (64.2). 119SnNMR (CDCl3) δ: -7.39 ppm. Anal. Calcd. for C50H74O4Sn2: C, 61.50, H, 7.64. Found: C, 61.64, H, 7.70.
2.1.2. (S)-1,1'-binaphthalene-2,2'-diyl-bis[3-(triphenylestannyl)propanoate] (S)-4
The product (S)-4 was eluted with hexane/ethyl acetate (90:10) as colourless oil in a 86% yield. [α]D25 -11.09 (c=0.82, CHCl3). 1HNMR (300 MHz, CDCl3) δ: 1.41 (t, 2J(H, Sn)= 61.8, Hz, 3J(H,H)= 7.42 Hz, 2 x CH2, 4H), 2.51 (t, 3J(H,H)= 7.42 Hz, 2 x CH2, 4H), 7.23 (d, 3J(H,H)= 8.9 Hz, Ar-H, 2H), 7.36-7.45 (m, Ar-H, 4H), 7.46-7.58 (m, Ar-H, 20H), 7.62-7.65 (m, Ar-H, 12H), 7.97-8.00 (m, Ar-H, 4H). 13CNMR (75.4 MHz, CDCl3) δ: 5.02 (390.1), 29.34 (18.4), 122.03, 123.44, 125.35, 125.72, 126.13, 126.74, 128.54, 128.85, 128.97, 129.06, 131.48, 133.36, 137.06 (35.3), 137.48, 138.28 (504.5), 146.81, 171.42 (64.9). 119SnNMR (CDCl3) δ: -99.05 ppm. Anal. Calcd. for C62H50O4Sn2: C, 67.91, H, 4.60. Found: C, 68.05, H, 4.67.
2.1.3. 1,1'-binaphthalene-2,2'-diyl-bis[2-methyl-3-(tri-n-buthylestannyl)propanoate] (5a-d, 5a-b)
The mixture of 5a-d was eluted with hexane/ethyl ether (97:3) as a white solid in a 67% yield. By re-chromatography, the diastereomers 5a-b were eluted with hexane/ethyl ether (96:4) as a white solid in a 34% yield in a 61:39 ratio [d.e.: 22%]. 1HNMR (300 MHz, CDCl3) δ: 0.46 (dd, 3J(H,H)= 6.9 Hz, 2J(H,H)= 2.3 Hz, 4 x CH2, 8H), 0.60-0.67 (m, 12 x CH2, 24H), 0.81 (t, 3J(H,H)= 7.3 Hz, 12 x CH3, 36H), 1.11-1.22 (m, 4CH3 y 12CH2, 36H), 1.23-1.33 (m, 12 x CH2, 24H), 2.23-2.42 (m, 4 x CH, 4H), 7.21-4.23 (m, Ar-H, 8H), 7.28-7.42 (m, Ar-H, 8H), 7.77-7.93 (m, Ar-H, 8H). 13CNMR (75.4 MHz, CDCl3) δ: 9.39 (309.9), 9.41 (323.6), 12.27 (281.5), 13.84, 19.73 (22.8), 27.32 (55.5), 29.20 (17.0), 37.30 (23.6), 37.33 (20.2), 122.11, 123.81, 123.83, 125.70, 126.27, 126.76, 127.93, 129.30, 129.34, 131.59, 131.61, 133.56, 133.60, 147.02, 175.91, 175.92. 119SnNMR (CDCl3) δ: -11.34, -11.82. Anal. Calcd. for C52H78O4Sn2: C, 62.17, H, 7.83. Found: C, 62.32, H, 7.89.
2.1.4. 1,1'-binaphthalene-2,2'-diyl-bis[2-methyl-3-(triphenylestannyl)propanoate] (6a-d)
The mixture of 6a-d was eluted with hexane/ethyl acetate (95:5) as a white solid in a 84% yield. 1HNMR (300 MHz, CDCl3) δ: 0.42-0.60 (m, 8 x CH3, 24H), 0.84-1.08 (m, 8 x CH2, 16H), 2.39-2.66 (m, 8 x CH, 8H), 6.92-7.12 (m, Ar-H, 16H), 7.38-7.55 (m, Ar-H, 64H), 7.59-7.70 (m, Ar-H, 56H), 7.84-7.93 (m, Ar-H, 32H). 13CNMR (75.4 MHz, CDCl3) δ: 13.59, 13.62, 13.70, 13.72, 18.29, 35.34, 35.29, 35.27, 35.23, 120.22, 120.25, 120.29, 120.31, 121.72, 121.75, 123.90, 123.94, 124.25, 124.26, 124.36, 124.90, 124.92, 126.13, 126.18, 126.41, 126.39, 126.74, 126.94, 126.99, 127.06, 127.11, 127.13, 129.68, 129.71, 129.73, 131.54, 131.56, 131.61, 131.62, 153..33, 135.71, 136.98, 137.01, 137.03, 137.36, 144.95, 144.09, 145.01, 173.29, 173.34, 173.38, 173.41. 119SnNMR (CDCl3) δ: -103.24, -103.61, -103.74, -103.91. Anal. Calcd. for C64H54O4Sn2: C, 68.36, H, 4.84. Found: C, 68.53, H, 4.94.
2.2. General Procedure for the Addition of di-n-butyltin Chlorohydride to BINOL Unsaturated Diesters Initiated with AIBN at 40°C (METHOD A) [17Gerbino, D.C.; Scoccia, J.; Koll, L.C.; Mandolesi, S.D.; Podestá, J.C. Stereoselective synthesis and some properties of new chlorodiorganotin-substituted macrodiolides. Organometallics, 2012, 31, 662-671.
[http://dx.doi.org/10.1021/om200987t] ]
Di-n-butyltin dichloride (0.18 g, 0.61 mmoles) was dissolved in dry toluene (3.4 mL), and di-n-butyltin dihydride (0.12 mL, 0.14 g, 0.61 mmoles) was added. The mixture was stirred for about 30 min, with monitoring the reaction by IR spectroscopy to verify the formation of di-n-butyltin chlorohydride. Then, a solution of diester (0.51 mmoles) in dry toluene (5.7 mL) was added with a syringe slowly to the mixture together with a catalytic amount of azo-bis-isobutyronitrile (AIBN) as a radical initiator. The reaction mixture was heated to 40° C and was monitored by TLC and IR spectroscopy. After one hour, the solvent was then distilled off under reduced pressure. The crude product thus obtained was directly purified by column chromatography using silica gel 60.
2.3. General Procedure for the Addition of Diphenyltin Chlorohydride to BINOL Unsaturated Diesters Initiated with AIBN at 40°C (METHOD A) [17Gerbino, D.C.; Scoccia, J.; Koll, L.C.; Mandolesi, S.D.; Podestá, J.C. Stereoselective synthesis and some properties of new chlorodiorganotin-substituted macrodiolides. Organometallics, 2012, 31, 662-671.
[http://dx.doi.org/10.1021/om200987t] ]
Diphenyltin dichloride (0.21 g, 0.61 mmoles) was dissolved in dry toluene (1 mL), and diphenyltin dihydride (0.12 mL, 0.17 g, 0.61 mmoles) was added. The mixture was stirred for about 1 hour, monitoring the reaction by IR spectroscopy to verify the formation of diphenyltin chlorohydride. Then, a solution of diester (0.51 mmoles) in dry toluene (5.7 mL) was added with a syringe slowly to the mixture together with a catalytic amount of azo-bis-isobutyronitrile (AIBN) as a radical initiator. The reaction mixture was heated to 40°C and was monitored by TLC and IR spectroscopy. After one hour, the solvent was then distilled off under reduced pressure. The crude product thus obtained was directly purified by column chromatography using silica gel 60.
2.4. Procedure for the Addition of di-n-butyltin Chlorohydride to BINOL Dimethacrylate Diesters at −78°C Initiated with Et3B (METHOD B) [17Gerbino, D.C.; Scoccia, J.; Koll, L.C.; Mandolesi, S.D.; Podestá, J.C. Stereoselective synthesis and some properties of new chlorodiorganotin-substituted macrodiolides. Organometallics, 2012, 31, 662-671.
[http://dx.doi.org/10.1021/om200987t] , 34Ieki, R.; Kani, Y.; Tsunoi, S.; Shibata, I. Transition-metal-free coupling reaction of vinylcyclopropanes with aldehydes catalyzed by tin hydride. Chemistry, 2015, 21(16), 6295-6300.
[http://dx.doi.org/10.1002/chem.201406496] [PMID: 25753150] ]
Di-n-butyltin dihydride (0.12 mL, 0.14 g, 0.61 mmoles) was added to a solution of di-n-butyltin dichloride (0.18 g, 0.61 mmoles) in dry toluene (3.4 mL) at room temperature. After being stirred for 30 min, the formation of the di-n-butyltin chlorohydride was verified by IR and the mixture was cooled to −78°C. A solution of diester (S)-1b (0.21 g, 0.51 mmoles) in dry toluene (5.7 mL) was added with a syringe slowly to the mixture. Immediately, Et3B (0.10 mmoles, 0.0099 g, 0.011 mL) was added and the mixture was stirred at −78°C for 8 h with monitoring by TLC and IR spectroscopy. The solvent was distilled off under reduced pressure.
2.5. Procedure for the Addition of Diphenyltin Chlorohydride to BINOL Dimethacrylate Diesters at −78°C Initiated with Et3B (METHOD B) [17Gerbino, D.C.; Scoccia, J.; Koll, L.C.; Mandolesi, S.D.; Podestá, J.C. Stereoselective synthesis and some properties of new chlorodiorganotin-substituted macrodiolides. Organometallics, 2012, 31, 662-671.
[http://dx.doi.org/10.1021/om200987t] , 34Ieki, R.; Kani, Y.; Tsunoi, S.; Shibata, I. Transition-metal-free coupling reaction of vinylcyclopropanes with aldehydes catalyzed by tin hydride. Chemistry, 2015, 21(16), 6295-6300.
[http://dx.doi.org/10.1002/chem.201406496] [PMID: 25753150] ]
Diphenyltin dihydride (0.11 mL, 0.16 g, 0.57 mmoles) was added to a solution of diphenyltin dichloride (0.19 g, 0.57 mmoles) in dry toluene (0.5 mL) at room temperature. After being stirred for 1 hour, the formation of diphenyltin chlorohydride was verified by IR and the mixture was cooled down to −78°C. A solution of diester (0.47 mmoles) in dry toluene (4.5 mL) was added with a syringe slowly to the mixture. Immediately, Et3B (0.094 mmoles, 0.0092 g, 0.010 mL) was added and the mixture was stirred at −78°C for 10 h with monitoring by TLC and IR spectroscopy. The solvent was distilled off under reduced pressure.
2.5.1. (S)-1,1'-binaphthalene-2,2'-diyl-bis[3-(chlorodi-n-buthylestannyl)propanoate] (S)-8 Method A
The product (S)-8 was eluted with hexane/ethyl acetate (98:2) as a yellow oil in a 65% yield. [α]D25 +170.80 (c=0.020, CHCl3). 1HNMR (300 MHz,CDCl3) δ: 0.77 (dd, 3J(H,H)= 7.3 Hz, 2CH2, 4H), 0.83 (dd, 3J(H,H)= 7.3 Hz, 2CH2, 4H), 1.06-1.39 (m, 4 x CH3, 4 x CH2, 20H), 1.43-4.56 (m, 4 x CH2, 8H), 2.22-2.33 (m, 2 x CH2, 4H), 2.49-2.60 (m, 2 x CH2, 4H), 7.02-7.33 (m, Ar-H, 6H), 7.43 (dt, 3J(H,H)= 8.2 Hz, 4J(H,H)= 1.8 Hz, Ar-H, 2H), 7.89 (d, 3J(H,H)= 8.2 Hz, Ar-H, 2H), 7.43 (d, 3J(H,H)= 8.2 Hz, Ar-H, 2H). 13CNMR (75.4 MHz, CDCl3) δ: 11.59 (388.6), 13.81, 18.11 (647.0), 18.80, 26.88 (85.1), 27.86 (41.1), 29.96 (27.2), 120.83, 123.06, 126.24, 126.46, 127.40, 128.39, 130.48, 131.92, 133.06, 146.34, 178.77 (24.4). 119SnNMR (CDCl3) δ: 85.88 ppm. Anal. Calcd. for C42H56Cl2O4Sn2: C, 54.06, H, 6.05. Found: C, 54.24, H, 6.12.
2.5.2. (S)-1,1'-binaphthalene-2,2'-diyl-bis[3-(chlorodiphenylestannyl)propanoate] (S)-9 Method A
The product (S)-9 was eluted with hexane/ethyl acetate (98:2) as a yellow oil in a 95% yield. [α]D25 +36.32 (c=0.038, CHCl3). 1HNMR (300 MHz, CDCl3) δ: 1.26-1.30 (m, CH2, 2H), 1.30-1.40 (m, CH2, 2H), 2.16-2.28 (m, 2 x CH2, 4H), 6.83-7.07 (m, Ar-H, 8H), 7.21-7.42 (m, Ar-H, 20H), 7.82-7.89 (m, Ar-H, 4H). 13CNMR (75.4 MHz, CDCl3) δ: 13.04 (513.0), 29.38 (32.8), 120.72, 122.57, 125.30, 125.64, 126.38, 127.31, 128.22, 128.58, 129.10, 129.17, 129.65, 130.14, 130.49, 131.66, 132.723, 136.11 (45.3), 146.13, 179.36 (43.6). 119SnNMR (CDCl3) δ: -52.55. Anal. Calcd. for C50H40Cl2O4Sn2: C, 59.27, H, 3.98. Found: C, 59.45, H, 4.07.
2.5.3. 1,1'-binaphthalene-2,2'-diyl-bis[3-(chlorodi-n-buthylestannyl)-2-methyl propanoate] (10a-d) Method A
The mixture of diastereoisomers 10a-d was isolated by column chromatography using silica gel 60 as the stationary phase eluting as a yellow oil with a mixture of hexane: ethyl acetate (90:10) in a 78% yield. Method B. The mixture of diastereoisomers 10a-d was isolated by column chromatography using silica gel 60 as the stationary phase eluting as a yellow oil with a mixture of hexane: ethyl acetate (90:10) in a 40% yield. 1HNMR (300 MHz, CDCl3) δ: 0.18 (d, 3J(H,H)= 7.2 Hz, 2 x CH3, 6H), 0.30 (d, 3J(H,H)= 7.2 Hz, 2 x CH3, 6H), 0.59 (d, 3J(H,H)= 7.2 Hz, 2 x CH3, 6H), 0.76-0.91 (m, 2CH3, 4 x 2CH2, 22H), 1.04-1.84 (m, 4 x 12CH2, 4 x 4CH3, 144H), 2.37-2.91 (m, 4 x 2CH, 8H), 7.04-7.32 (m, Ar-H, 24H), 7.40-7.45 (m, Ar-H, 8H); 7.86-7.97 (m, Ar-H, 16H). 13CNMR (75.4 MHz, CDCl3) δ: 12.68, 18.33, 18.63, 18.90, 19.00, 20.55, 20.56, 20.73, 20.85, 25.43, 25.57, 26.08, 26.28, 31.27, 31.87, 35.55, 35.62, 35.66, 35.74, 119.65, 121.97, 124.74, 125.32, 126.22, 127.20, 129.07, 130.71, 131.82, 145.12, 180.03, 180.28, 180.35, 180.61. 119SnNMR (CDCl3) δ: 72.95, 70.09, 69.68, 68.72. Anal. Calcd. for C44H60Cl2O4Sn2: C, 54.98, H, 6.29. Found: C, 55.12, H, 6.35.
2.5.4. 1,1'-binaphthalene-2,2'-diyl-bis[3-(Chlorodiphenylestannyl)-2-methyl Propanoate] (11a-d) Method A
The mixture of diastereoisomers 11a-d was purified by column chromatography using silica gel 60 as the stationary phase eluting as a yellow oil with a mixture of hexane: ethyl acetate (98:2) in a 96% yield. Method B. The mixture of diastereoisomers 11a-d was isolated by column chromatography using silica gel 60 as the stationary phase eluting as a yellow oil with a mixture of hexane: ethyl acetate (98:2) in a 65% yield. 1HNMR (300 MHz, CDCl3) δ: 0.04 (d, 3J(H,H)= 7.2 Hz, 2 x CH3, 6H), 0.19 (d, 3J(H,H)= 7.2 Hz, 2 x CH3, 6H), 0.28 (d, 3J(H,H)= 7.2 Hz, 2 x CH3, 6H), 0.44 (d, 3J(H,H)= 7.2 Hz, 2 x CH3, 6H), 0.89-1.34 (m, 4 x 2CH2, 16H), 2.32-2.78 (m, 4 x 2CH, 8H), 6.61-7.22 (m, Ar-H, 112H), 7.78-7.88 (m, Ar-H, 16H). 13CNMR (75.4 MHz, CDCl3) δ: 18.78, 19.08, 19.18, 19.32, 23.32, 23.40, 36.45, 36.53, 36.60, 36.73, 118.77, 118.93, 119.00, 120.74, 120.94, 121.28, 123.60, 123.71, 123.78, 123.83, 124.63, 124.73, 124.78, 125.60, 125.70, 126.81, 127.42, 127.85, 128.26, 128.75, 129.91, 129.99, 130.11, 130.99, 131.01, 131.08, 134.41, 144.20, 144.29, 144.31, 144.36, 181.25, 181.63, 181.75, 181.85. 119SnNMR (CDCl3) δ: -63.69, -66.73, -68.68, -70.39. Anal. Calcd. for C52H44Cl2O4Sn2: C, 59.98, H, 4.26. Found: C, 60.12, H, 4.34.
2.6. Biology, Antifungal Evaluation, Microorganisms and Media
For the antifungal evaluation, the standardized strains C. albicans ATCC 10231 and C. neoformans ATCC 32264 from the American Type Culture Collection (ATCC), Rockville, MD, USA were used. Strains were grown on Sabouraud-chloramphenicol agar slants for 48 h at 30°C, maintained on slopes of Sabouraud-dextrose Agar (SDA, Oxoid), and subcultured every 15 days to prevent pleomorphic transformations. Inocula of the cell or spore suspensions were obtained according to reported procedures and adjusted to 1-5x 103 cells/spores with colony forming units (CFU)/mL.
2.7. Antifungal Susceptibility Testing
Minimum Inhibitory Concentration (MIC) of each extract or compound was determined by using broth microdilution techniques according to the guidelines of the National Committee for Clinical Laboratory Standards for yeasts (M27-A3). MIC values were determined in RPMI-1640 (Sigma-Aldrich, St Louis, MO, USA), buffered to pH 7.0 with MOPS. Microtiter trays were incubated at 35°C in a moist, dark chamber, and MICs were visually recorded at 48 h. For the assay, stock solutions of pure compounds were two-fold diluted with RPMI from 256 to 3.9 µg/mL (final volume = 100 µL) and a final DMSO concentration ≤ 1%. A volume of 100 µL of inoculum suspension was added to each well with the exception of the sterility control where sterile water was added to the well instead. Amphotericin B (Sigma Aldrich) was used as a positive control. Endpoints were defined as the lowest concentration of drug resulting in total inhibition (MIC) of visual growth compared to the growth in the control wells containing no antifungal. The IC50 was defined as the lowest concentration of a compound that produced 50% reduction in the growth control (culture media with the microorganism but without the addition of any compound), and was determined spectrophotometrically with the aid of a VERSA Max microplate reader (Molecular Devices, Sunnyvale, CA, USA) and the % of inhibition for each compound at the different concentrations was calculated as follows: 100x (OD (optical density) 405 MTW - OD 405 SCW)/(OD 405 GCW - OD 405 SCW). Tests were conducted in triplicate.
2.8. X-ray Diffraction Analysis
The X-ray data were collected at 293(2) K with a Nonius Kappa-CCD with Mo source diffractometer. The used software for data collection included: APEX3: Bruker (2017). APEX3. Bruker AXS Inc., Madison, Wisconsin, USA; for structure solution SHELXT [29Sheldrick, G.M. SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv., 2015, 71(Pt 1), 3-8.
[http://dx.doi.org/10.1107/S2053273314026370] [PMID: 25537383] ] and for structure refinement SHELXL [30Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem., 2015, 71(Pt 1), 3-8.
[http://dx.doi.org/10.1107/S2053229614024218] [PMID: 25567568] ]. All H atoms bonded to carbon were placed with idealized geometry and refined using riding model with C--H = 0.95 Å, Uiso(H) = 1.2 Ueq(C) for CH. The absolute configuration was determined by the Flack parameter and was assumed to be unchanged during the reaction and was not determined by diffraction methods. The structural data have been deposited with Cambridge Crystallographic Data Centre, CCDC number CCDC 1589534. Crystallographic data of (S)-1e for the structural analyses have been deposited with the Cambridge Crystallographic Data Centre, CCDC 1589534. Copies of this information may be obtained free of charge from the Director, CCDC, 12 Union Road, Cambridge, CB21EZ, UK, fax: +44 1223 336 033; email deposit@ccdc.cam.ac.uk or www:http:// www.ccdc.cam.ac.uk.
3. RESULTS AND DISCUSSION
Based on our previous results [16Gerbino, D.C.; Koll, L.C.; Mandolesi, S.D.; Podestá, J.C. Stereoselective radical tandem cyclo hydrostannation of optically active di-unsaturated esters of TADDOL. Organometallics, 2008, 27, 660-665.
[http://dx.doi.org/10.1021/om700693p] -18Zacconi, F.C.; Ocampo, R.A.; Podestá, J.C.; Koll, L.C. Synthesis of organotin substituted tricyclic macrodiolides. J. Braz. Chem. Soc., 2016, 27, 484-492.], we assumed that the radical hydrostannation of (S)-BINOL unsaturated diesters (S)-1a-e could lead to additional products to one or both olefinic systems (compounds type A or B, Scheme 1) or the corresponding cycloadducts (compounds type C, Scheme 1).
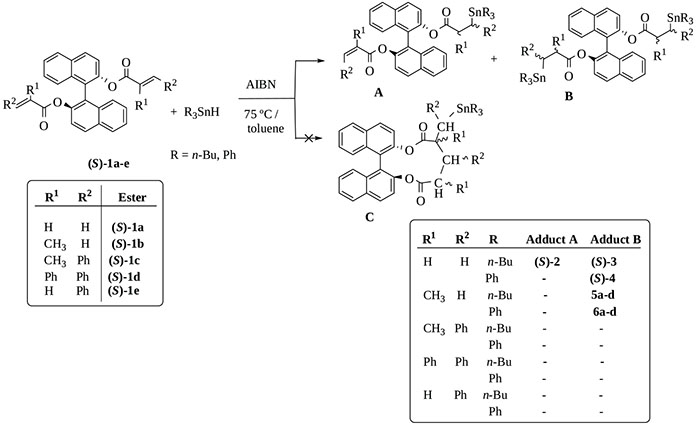 |
Scheme (1) Radical addition of triorganotin hydrides to (S)-BINOL unsaturated diesters. |
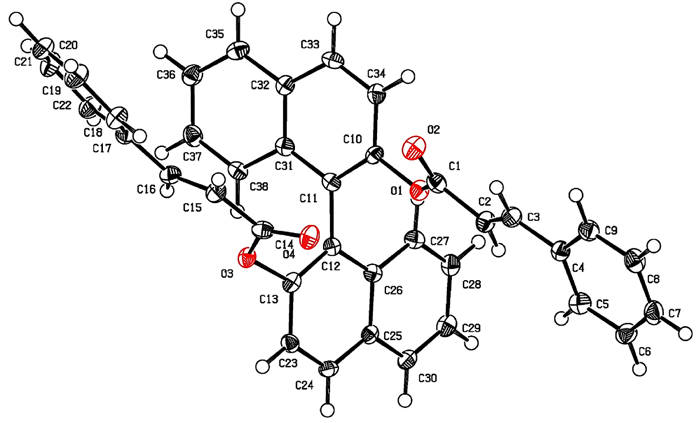 |
Fig. (3) X-ray structure of diester (S)-1e. |
After performing the reaction between (S)-1,1'-binaphtyl-2-2'-diyl-diacrylates ((S)-1a-e) and the corresponding tri-n-butyl- and triphenyltin hydrides under already known radical conditions [16Gerbino, D.C.; Koll, L.C.; Mandolesi, S.D.; Podestá, J.C. Stereoselective radical tandem cyclo hydrostannation of optically active di-unsaturated esters of TADDOL. Organometallics, 2008, 27, 660-665.
[http://dx.doi.org/10.1021/om700693p] -18Zacconi, F.C.; Ocampo, R.A.; Podestá, J.C.; Koll, L.C. Synthesis of organotin substituted tricyclic macrodiolides. J. Braz. Chem. Soc., 2016, 27, 484-492., 31Leusink, A.J.; Noltes, J.G. Studies in group IV organometallic chemistry XXXI. Organotin hydride adducts with tin atoms in α,β- or β,β-positions. J. Organomet. Chem., 1969, 16, 91-102.
[http://dx.doi.org/10.1016/S0022-328X(00)81639-3] ], (anhydrous toluene, 1:1,2 substrate/ organotin hydride relation, inert atmosphere, 75°C and AIBN as radical initiator) only mixtures of type A and/or B adducts were formed when substrates (S)-1a and (S)-1b were employed. No reaction was observed with (S)-1c to (S)-1e, and the expected cyclized structure C was not observed in any case. The (S)-BINOL diester spatial arrangement could explain these results. This molecule, with C2 symmetry, adopts preferably the transoid conformation, where the bulky substituents located in positions 2 and 2' mutually move away. Thus, the olefinic groups are arranged opposite to the other and, therefore, the formation of the macrocycle is disfavored. This structural arrangement was confirmed through X-ray diffraction analysis of dicinnamate (S)-1e which produced suitable crystals that would confirm this approach (Fig. 3 ). Dicinnamate (S)-1e was obtained following the procedure of reference 13Iqbal, H.; Ali, S.; Shahzadi, S. Antituberculosis study of organotin(IV) complexes: A review. Cogent Chem., 2015, 1, 1029039.
). Dicinnamate (S)-1e was obtained following the procedure of reference 13Iqbal, H.; Ali, S.; Shahzadi, S. Antituberculosis study of organotin(IV) complexes: A review. Cogent Chem., 2015, 1, 1029039.
[http://dx.doi.org/10.1080/23312009.2015.1029039] . (See Supporting Information for X-Ray diffraction analysis). Crystal structural data was deposited at the Cambridge Crystallographic Data Centre with Deposition number: CCDC 1589534.
Increasing the proportion of organotin hydride, the optimal substrate / R3SnH relation determined in order to obtain a higher proportion of type B bis-trialkyltin addition products was 1:2,4 under the previously mentioned conditions. TLC chromatographic monitoring of the reaction showed that they were completed after one hour. Electronic and steric factors usually determine the rate of addition to olefins and in both cases, the alkene has the ester group as an activating electron-withdrawing group and the β-carbon is unsubstituted so there is no steric hindrance for the regioselective binding of the trialkylstannyl group to the terminal carbon. However, this last factor would affect diesters (S)-1c to (S)-1e, which remained unreacted after 10 h. Table 1 summarizes the results obtained in the radical hydrostannation of the reactive substrates (S)-1a and (S)-1b.
The reaction with Bu3SnH with the diester (S)-1a, gave a 89% mixture of mono- and di-addition products (S)-2 and (S)-3 in a 31:69 proportion respectively which could be separated by chromatography although with low final yield of pure bis-stannylated adduct (S)-3 (Table 1, entry 1, 67%, Supporting Information, Fig. S1). Increasing the substrate/Bu3SnH relation to 1:3 provides a 20:80 mixture of single (A) and double addition products (B) but in lower yield (63%) from which (S)-3 was obtained in a 45% yield. Type A adduct (S)-2 could not be isolated in a pure form so it could not be characterized. The same organotin hydride reacted with substrate (S)-1b to yield in this case only bis-addition adducts 5a-d, obtained as a mixture of four diastereomers in relation 50:13:25:12 (Table 1, entry 3, 67%) from which it was possible to separate two of them by re-chromatography, named 5a-b (34%), 119SnNMR (δ, ppm) -11.34 and -11.82 (see Supporting Information, Fig. S2 and experimental section).
The best yields were observed in the reaction with Ph3SnH and only type B products were obtained. This can be attributed to the higher reactivity and higher hydrogen donating capability of Ph3SnH relative to Bu3SnH. (S)-4 adduct was obtained as a single enantiomer in 86% yield after purification (Table 1, entry 2; Supporting Information, Fig. S3), and 6a-d were obtained in 84% yield, as a mixture of four diastereomers in 8:19:42:31 relation, that could not be separated (Table 1, entry 4; Supporting Information, Fig. S4). All the obtained double addition products were purified by chromatography and characterized by 1H, 13C and 119SnNMR. In all cases, the diastereomeric composition (% D) was determined from the 119SnNMR spectra of the crude reaction products. Analysis of 1H and 13CNMR spectral data (Supporting Information, Tables S1-S4 and experimental section) confirms the structure of type B adducts (S)-3, (S)-4, 5a-b and 6a-d, showing in the 13CNMR spectrum only one signal with 3J(Sn,C) coupling constant, corresponding to C=O bond, for each stereoisomer.
Changing the organic ligand by halogen in the organotin hydride determines novel derivatives with the general structure of bis-halodiorganotin adduct and the differentiation of certain molecular properties that would help further studies thereof [32Mitchell, T.N.; Podestá, J.C.; Ayala, A.; Chopa, A.B. Experimental test of karplus-type relationships for 3J(Sn, C) and 3J(Sn, H). Magn. Reson. Chem., 1988, 26, 497-500.
[http://dx.doi.org/10.1002/mrc.1260260612] -34Ieki, R.; Kani, Y.; Tsunoi, S.; Shibata, I. Transition-metal-free coupling reaction of vinylcyclopropanes with aldehydes catalyzed by tin hydride. Chemistry, 2015, 21(16), 6295-6300.
[http://dx.doi.org/10.1002/chem.201406496] [PMID: 25753150] ].
As it is already known, a halogen atom as a ligand bound to tin renders the latter more electropositive, which favors its intramolecular coordination with the carbonyl in β- respective to the ester group through a five-member cycle (Fig. 4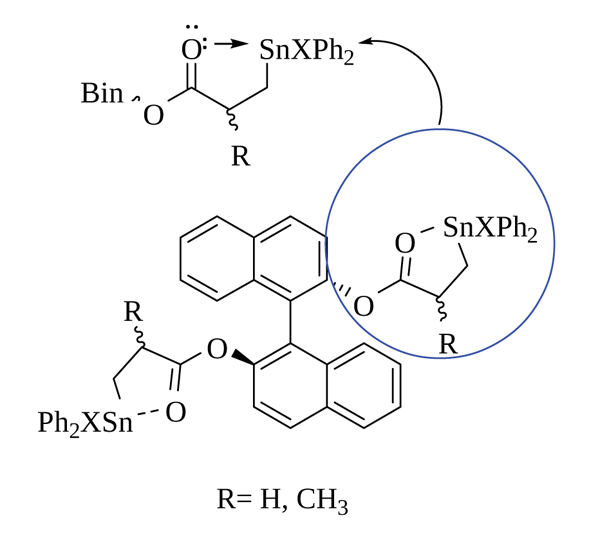 ) which could determine a particular spectroscopic behavior [35Podestá, J.C.; Chopa, A.B.; Ayala, A.D.; Koll, L.C. Organotin compounds IX. Reactions between trialkylstannyl-substituted esters and trimethylsilyl halides: Exchange vs. hydrolysis. J. Organomet. Chem., 1987, 333, 25-36.].
) which could determine a particular spectroscopic behavior [35Podestá, J.C.; Chopa, A.B.; Ayala, A.D.; Koll, L.C. Organotin compounds IX. Reactions between trialkylstannyl-substituted esters and trimethylsilyl halides: Exchange vs. hydrolysis. J. Organomet. Chem., 1987, 333, 25-36.].
Haloorganotin derivatives of this type could be obtained through radical addition of mixed organotin halohydrides R2SnHX. These highly reactive tin hydrides undergo radical chain reactions at low temperature (−78°C to 40ºC) not higher than 40°C in order to prevent their decomposition. Their higher Lewis acidity (in comparison with the most commonly used hydrostannylation reagent Bu3SnH or Ph3SnH) leads to much better regio- and stereoselectivities.17 Considering the formation of an intermediate with intramolecular coordination during the mechanistic steps of the radical halohydrostannation, this could result in a higher stereoselectivity or differences in the course of the reaction.
We added dialkyltin chlorohydrides R2SnHCl (R= n-Bu, Ph) to the (S)-BINOL unsaturated diesters (S)-1a and (S)-1b, and then analyzed whether substantial changes in diastereoselectivity and yield were produced in the products obtained (Scheme 2). The mixed hydrides were prepared by exchange reactions between an equimolecular mixture of R2SnH2 and R2SnCl2 and used in-situ [36Thiele, C.M.; Mitchell, T.M. Hydrostannylation of propargylic alcohols using mixed tin hydrides. Eur. J. Org. Chem., 2004, 2, 337-353.
[http://dx.doi.org/10.1002/ejoc.200300489] ]. The halohydrostannations were carried out under inert atmosphere and radical conditions, using AIBN as radical initiator, using 2.4 equivalents of R2SnHCl in toluene and keeping the mixture at room temperature (Method A) [37Podestá, J.C.; Chopa, A.B. Organotin compounds III. Organotin chlorohydride additions to methyl e-disubstituted propenoates. J. Organomet. Chem., 1982, 229, 223-228.
[http://dx.doi.org/10.1016/S0022-328X(00)83815-2] ].

 |
Fig. (4) Five-member cycle intramolecular coordination in β-haloorganotin esters. |
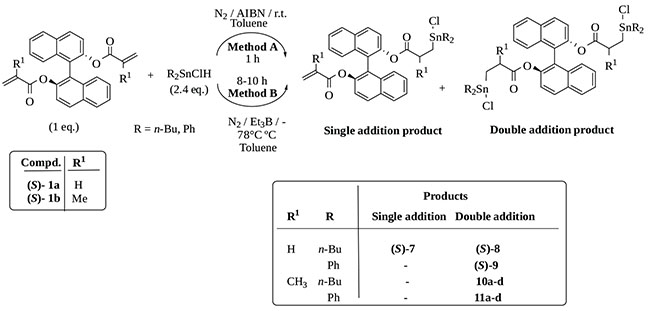 |
Scheme (2) Addition of dialkyltin chlorohydrides R2SnHCl to the unsaturated diesters (S)-1a and (S)-1b. |
Except for the addition of n-Bu2SnHCl to (S)-1a where a single addition product was detected (Table 2, entry 1), the reaction gave exclusively double addition products after 1 h. Yields were almost quantitative for Ph2SnHCl addition and smaller for n-Bu2SnHCl probably due to its lower reactivity (H donor capability). The diastereoisomeric mixtures 10a-d and 11a-d could not be separated and no improvement in diastereoselectivity was observed under these conditions.
Taking into account that the stereoselectivity in radical reactions may be improved by temperature lowering [17Gerbino, D.C.; Scoccia, J.; Koll, L.C.; Mandolesi, S.D.; Podestá, J.C. Stereoselective synthesis and some properties of new chlorodiorganotin-substituted macrodiolides. Organometallics, 2012, 31, 662-671.
[http://dx.doi.org/10.1021/om200987t] , 38Curran, D.P. Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications, chapter 1., 1996, ], we studied the chlorohydrostannations of 1b at -78ºC using Et3B as a radical initiator (Method B, Table 3) but without success. We observed longer reaction times, lower yields and no improvement in the diastereomeric relation. Spectroscopic characteristics of this bis-chloroorganotin adducts are reported in the Supporting Information, Tables S5-S8.
All the products obtained, either those enantiomerically pure (S)-3, (S)-4, (S)-8, (S)-9 or the diastereoisomeric mixtures 5a-d, 6a-d, 10a-d, 11a-d were tested for antifungal activity assays. Despite having separated two diastereoisomers (5a-b), the mixture of four (5a-d) was used in order to have a uniform comparison criterion to all diastereomeric mixtures.
4. ANTIFUNGAL ACTIVITY ASSAYS
Compounds S-3, S-4, S-8, S-9, 5a-d, 6a-d, 10a-d and 11a-d were tested for antifungal properties against C. albicans ATCC 10231 and C. neoformans ATCC 32276 (Table 4). To assess antifungal activities, the broth microdilution method M27-A3 for yeasts of the Clinical and Laboratory Standards Institute was used [39CLSI. Reference method for broth dilution antifungal susceptibility testing of yeasts; Approved standard, CLSI Document M27 A3. 2008, ].
The percentage of inhibition of each fungus was determined for each compound at two-fold dilutions in the range 250-3.9-µg/mL where MIC and IC50 (the minimum concentration that inhibits 100 and 50% of fungal growth) are shown in the rightmost columns. Amphotericin B (Sigma Aldrich, St Louis, MO, USA) was used as a positive control.
The results obtained in the antifungal assays against C. albicans and C. neoformans (Table 4) allowed us to observe some trends in the structural-activity relationships: (a) There is not a clear difference in activity of the compounds against C. albicans and C. neoformans in the case of (S)-3, (S)-4, 6a-d and 5a-d. For example, it can be observed that the diastereomers 6a-d possess an IC50 of 1µg/mL against both the species, considering that one dilution is the error of the method [40Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother., 2003, 52(1), 1.
[http://dx.doi.org/10.1093/jac/dkg301] [PMID: 12805255] ]. Stereoisomers (S)-3, (S)-4, and 5a-d gave similar values of IC50 = 250 µg/mL. However, the behavior of (S)-9, 11a-d, (S)-8 and 10a-d is different. It can be observed that the activity of the enantiomeric form (S)-9, while is important against both strains, is lower for C. albicans than for C. neoformans (IC50 = 3.9 µg/mL and 1.9 µg/mL respectively) while in the case of diastereomers 11a-d and enantiomer (S)-8, the IC50 values are not so good (31.25 and 15.6 µg/mL; 62.5 and 125 µg/mL). In turn, diastereoisomers 10a-d showed a very good IC50 value of 2 µg/mL for C. albicans but low activity for C. neoformans (IC50 = 15.6 µg/mL).(For the sake of clarity see Fig. (5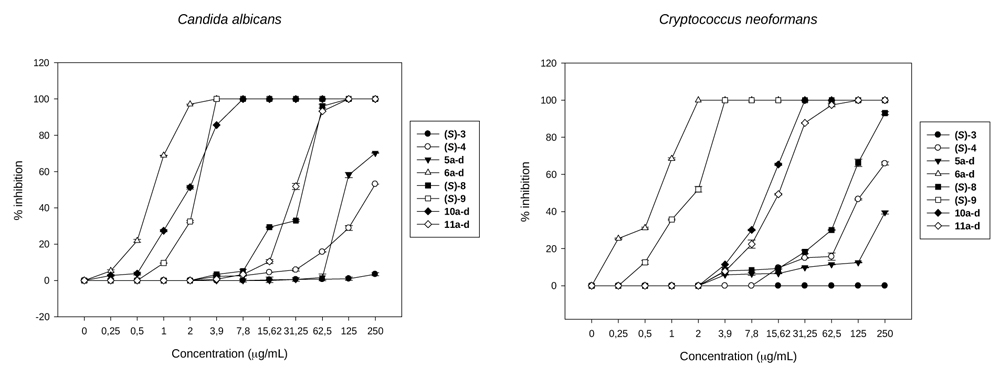 ). A and B). (b) If the comparisons are made taking into account different organic ligands on the Sn atom, there are clear differences in the activity: (b1) most compounds with Ph3Sn- substituents like 6a-d, with IC50 = 1 µg/mL for both fungi (except for (S)-4 that posseses IC50 = 250 µg/mL) are highly active compared with those possessing n-Bu3Sn- (5a-d, (S)-3, IC50 = 250 and >250 µg/mL respectively for both fungi). (b2) A methyl group in the alpha position (and therefore the introduction of a new chiral center leading to diastereomers) shows that the methyl group does not play a clear role in the antifungal activity. For example, 6a-d possess higher activity than (S)-4, whereas 11a-d show lower activities than (S)-9. (c) In contrast with point (b), there are clear differences in activity when comparing compounds with different substituents on the Sn atom: (c1) from the compounds containing SnPh3 (6a-d and (S)-4), 6a-d is highly active (IC50 values = 1 µg/mL for both fungi) compared with those possessing SnBu3 ((S)-3 and 5a-d which showed IC50 = 250 - > 250 µg/mL for both fungi); (c2) When one phenyl of SnPh3 is changed into Cl leading to SnPh2Cl ((S)-9 and 11a-d) changes in the activity are observed: the IC50 of (S)-9 is much better than that observed for (S)-4. However, when 11a-d is compared with 6a-d, the antifungal effect is reversed, from an IC50 = 31.25 for C. albicans and 15.6 µg/mL for C. neoformans (11a-d) to an IC50 = 1 µg/mL for 6a-d in both fungi. (c3) When SnBu3 in (S)-3 and 5a-d is changed into SnBu2Cl, leading to (S)-8 and 10a-d respectively, the IC50 decreases from IC50 ≥ 250 in (S)-3 to 62.5-125 µg/mL in (S)-8 and from 250 µg/mL in 5a-d to 2-15.6 in 10a-d, the last one rendering a highly active compound. A comparative curve of the behavior of the most active structures against C. albicans and C. neoformans can be observed in Figs. (5 and 6
). A and B). (b) If the comparisons are made taking into account different organic ligands on the Sn atom, there are clear differences in the activity: (b1) most compounds with Ph3Sn- substituents like 6a-d, with IC50 = 1 µg/mL for both fungi (except for (S)-4 that posseses IC50 = 250 µg/mL) are highly active compared with those possessing n-Bu3Sn- (5a-d, (S)-3, IC50 = 250 and >250 µg/mL respectively for both fungi). (b2) A methyl group in the alpha position (and therefore the introduction of a new chiral center leading to diastereomers) shows that the methyl group does not play a clear role in the antifungal activity. For example, 6a-d possess higher activity than (S)-4, whereas 11a-d show lower activities than (S)-9. (c) In contrast with point (b), there are clear differences in activity when comparing compounds with different substituents on the Sn atom: (c1) from the compounds containing SnPh3 (6a-d and (S)-4), 6a-d is highly active (IC50 values = 1 µg/mL for both fungi) compared with those possessing SnBu3 ((S)-3 and 5a-d which showed IC50 = 250 - > 250 µg/mL for both fungi); (c2) When one phenyl of SnPh3 is changed into Cl leading to SnPh2Cl ((S)-9 and 11a-d) changes in the activity are observed: the IC50 of (S)-9 is much better than that observed for (S)-4. However, when 11a-d is compared with 6a-d, the antifungal effect is reversed, from an IC50 = 31.25 for C. albicans and 15.6 µg/mL for C. neoformans (11a-d) to an IC50 = 1 µg/mL for 6a-d in both fungi. (c3) When SnBu3 in (S)-3 and 5a-d is changed into SnBu2Cl, leading to (S)-8 and 10a-d respectively, the IC50 decreases from IC50 ≥ 250 in (S)-3 to 62.5-125 µg/mL in (S)-8 and from 250 µg/mL in 5a-d to 2-15.6 in 10a-d, the last one rendering a highly active compound. A comparative curve of the behavior of the most active structures against C. albicans and C. neoformans can be observed in Figs. (5 and 6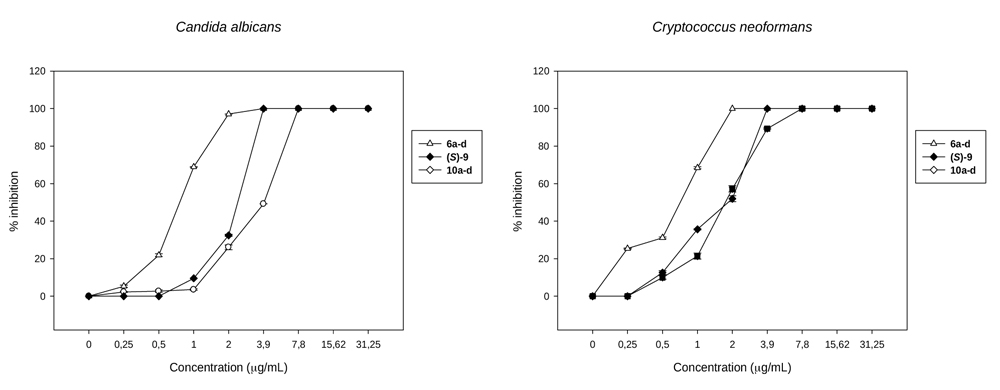 ).
).



CONCLUSION
After all these studies it could be assumed that the spatial disposition of the unsaturated ester groups linked to the C2 chiral core in (S)-BINOL diesters (S)-1a-e determines that the radical addition of organotin hydrides or organotin halohydrides leads to double addition adducts. From the nine pure organotin compounds synthesized here with defined stereochemistry, four of them ((S)-3, (S)-4, (S)-8 and (S)-9) are enantiomerically pure and four (5a-d, 6a-d, 10a-d and 11a-d) are diastereoisomeric mixtures. The antifungal properties were tested against two clinical important fungi with the diastereomeric mixture 6a-d, being the most active against both strains, followed by the enantiomer (S)-9 and the diastereomeric mixture 10a-d. Interesting enough, the active compounds behave similarly against C. albicans and C. neoformans and some of them possess IC values close to those of the reference drug. Regarding the most active structural features, these compounds contain preferably two or three phenyl groups joined to the tin atom bonded to an atropoisomeric core. Adducts 6a-d, with SnPh3 moiety, are highly active against both fungi compared with those possessing SnBu3 group.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Website along with the published article.
AVAILABILITY OF DATA AND MATERIALS
The structural data have been deposited with Cambridge Crystallographic Data Centre, CCDC number CCDC 1589534. Crystallographic data of (S)-1e for the structural analyses have been deposited with the Cambridge Crystallographic Data Centre, CCDC 1589534.
FUNDING
The authors acknowledge the sponsored grants of this work by the Consejo Nacional de Investigaciones y Técnicas (CONICET), Capital Federal, Argentina /PIP 112-201101- 00828, the Agencia Nacional de Promoción Científica y Tecnológica, (ANPCyT, Capital Federal, Argentina / PICT-2010-2644) and the Universidad Nacional del Sur (UNS, Bahía Blanca, Argentina). LS and SZ acknowledge ANPCyT, PICT2014-1170 and PICT2016-1833 for funds. LS and LK are members of the CONICET Researcher career. A fellowship from CONICET (Buenos Aires, Argentina) to ARC is gratefully acknowledged.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.
REFERENCES
| [1] | Hoch, M. Organotin compounds in the environment - An overview. Appl. Geochem., 2001, 16, 719-743. [http://dx.doi.org/10.1016/S0883-2927(00)00067-6] |
| [2] | Carraher, C.E. Organotin Polymers, in Macromolecules Containing Metal and Metal-Like Elements: Group IVA Polymers, chapter 10.2005, , 263-310. |
| [3] | Fent, K. Organotin in municipal wastewater and sewage sludge.Organotin: Environmental Fate and Effects., 1996, , 581-600. [http://dx.doi.org/10.1007/978-94-009-1507-7_28] |
| [4] | Cusack, P.A.; Hill, R. The Industrial Uses of Tin Chemicals., 1985, |
| [5] | Davies, A.G. Tin Chemistry: Fundamentals, Frontiers and Applications, 2008, |
| [6] | Smith, P.J., Ed.; Chemistry of Tin., (2nd ed. ), 1997, |
| [7] | Tarassoli, A.; Sedaghat, T. Organometallic Chemistry Research Perspectives, chapter 7.2007, , 221-248. |
| [8] | Gabriele, M.; Kuivila, H.G.; Cochran, J.C. Catalysis of the Diels-Alder reaction by organotin halides. Main Group Met. Chem., 1998, 21, 207-210. [http://dx.doi.org/10.1515/MGMC.1998.21.4.207] |
| [9] | Hoshi, T.; Shionoiri, H.; Katano, M.; Suzuki, T.; Hagiwara, H. Synthesis, structures and preliminary biological screening of bis(diphenyl)chlorotin complexes and adducts: Ph2ClSn–CH2–R–CH2–SnClPh2,R=p-C6H4,CH2CH2. Tetrahedron Asymmetry, 2002, 13, 2167-2175. [http://dx.doi.org/10.1016/S0957-4166(02)00578-5] |
| [10] | Takahashi, H.; Yasui, S.; Tsunoi, S.; Shibata, I. Catalytic cycloaddition of 2-methyleneaziridines with 1,1-dicyanoalkenes. Org. Lett., 2014, 16(4), 1192-1195. [http://dx.doi.org/10.1021/ol500062a] [PMID: 24495160] |
| [11] | Thoonen, S.H.L.; Deelman, B-J.; van Koten, G. Synthetic aspects of tetraorganotins and organotin(IV) halides. J. Organomet. Chem., 2004, 689, 2145-2157. [http://dx.doi.org/10.1016/j.jorganchem.2004.03.027] |
| [12] | Hadjikakou, S.K.; Hadjiliadis, N. Antiproliferative and anti-tumor activity of organotin compounds. Coord. Chem. Rev., 2009, 253, 235-249. [http://dx.doi.org/10.1016/j.ccr.2007.12.026] |
| [13] | Iqbal, H.; Ali, S.; Shahzadi, S. Antituberculosis study of organotin(IV) complexes: A review. Cogent Chem., 2015, 1, 1029039. [http://dx.doi.org/10.1080/23312009.2015.1029039] |
| [14] | Thodupunoori, S.K.; Alamudun, I.A.; Cervantes-Lee, F.; Gomez, F.D.; Carrasco, Y.P.; Pannell, K.H. Synthesis, structures and preliminary biological screening of bis(diphenyl)chlorotin complexes and adducts: Ph2ClSn-CH 2-R-CH2-SnClPh2, R = p-C6H4, CH2CH2. J. Organomet. Chem., 2006, 69, 11790-11796. [http://dx.doi.org/10.1016/j.jorganchem.2005.12.040] |
| [15] | Costantino, A.R.; Ocampo, R.A.; Montiel Schneider, M.G.; Fernandez, G.; Koll, L.C.; Mandolesi, S.D. Efficient routes to racemic and enantiomerically pure (S)-BINOL diesters. Synth. Commun., 2013, 43, 3192-3202. [http://dx.doi.org/10.1080/00397911.2013.774017] |
| [16] | Gerbino, D.C.; Koll, L.C.; Mandolesi, S.D.; Podestá, J.C. Stereoselective radical tandem cyclo hydrostannation of optically active di-unsaturated esters of TADDOL. Organometallics, 2008, 27, 660-665. [http://dx.doi.org/10.1021/om700693p] |
| [17] | Gerbino, D.C.; Scoccia, J.; Koll, L.C.; Mandolesi, S.D.; Podestá, J.C. Stereoselective synthesis and some properties of new chlorodiorganotin-substituted macrodiolides. Organometallics, 2012, 31, 662-671. [http://dx.doi.org/10.1021/om200987t] |
| [18] | Zacconi, F.C.; Ocampo, R.A.; Podestá, J.C.; Koll, L.C. Synthesis of organotin substituted tricyclic macrodiolides. J. Braz. Chem. Soc., 2016, 27, 484-492. |
| [19] | Laplante, S.R.; D Fader, L.; Fandrick, K.R.; Fandrick, D.R.; Hucke, O.; Kemper, R.; Miller, S.P.; Edwards, P.J. Assessing atropisomer axial chirality in drug discovery and development. J. Med. Chem., 2011, 54(20), 7005-7022. [http://dx.doi.org/10.1021/jm200584g] [PMID: 21848318] |
| [20] | McHenry, M.C.; Gavan, T.L. Vancomycin. Pediatr. Clin. North Am., 1983, 30(1), 31-47. [http://dx.doi.org/10.1016/S0031-3955(16)34318-8] [PMID: 6338468] |
| [21] | Keshmiri-Neghab, H.; Goliaei, B. Therapeutic potential of gossypol: an overview. Pharm. Biol., 2014, 52(1), 124-128. [http://dx.doi.org/10.3109/13880209.2013.832776] [PMID: 24073600] |
| [22] | Sprogøe, K.; Staek, D.; Ziegler, H.L.; Jensen, T.H.; Holm-Møller, S.B.; Jaroszewski, J.W. Combining HPLC-PDA-MS-SPE-NMR with circular dichroism for complete natural product characterization in crude extracts: levorotatory gossypol in Thespesia danis. J. Nat. Prod., 2008, 71(4), 516-519. [http://dx.doi.org/10.1021/np800010r] [PMID: 18290629] |
| [23] | Sawadjoon, S.; Kittakoop, P.; Kirtikara, K.; Vichai, V.; Tanticharoen, M.; Thebtaranonth, Y. Atropisomeric myristinins: selective COX-2 inhibitors and antifungal agents from Myristica cinnamomea. J. Org. Chem., 2002, 67(16), 5470-5475. [http://dx.doi.org/10.1021/jo020045d] [PMID: 12153244] |
| [24] | Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev., 2007, 20(1), 133-163. [http://dx.doi.org/10.1128/CMR.00029-06] [PMID: 17223626] |
| [25] | Butts, A.; Krysan, D.J. Antifungal drug discovery: something old and something new. PLoS Pathog., 2012, 8(9)e1002870 [http://dx.doi.org/10.1371/journal.ppat.1002870] [PMID: 22969422] |
| [26] | Gutch, R.S.; Nawange, S.R.; Singh, S.M.; Yadu, R.; Tiwari, A.; Gumasta, R.; Kavishwar, A. Antifungal susceptibility of clinical and environmental Cryptococcus neoformans and Cryptococcus gattii isolates in Jabalpur, a city of Madhya Pradesh in Central India. Braz. J. Microbiol., 2015, 46(4), 1125-1133. [http://dx.doi.org/10.1590/S1517-838246420140564] [PMID: 26691471] |
| [27] | Trpković, A.; Pekmezović, M.; Barać, A.; Crnčević Radović, L.; Arsić Arsenijević, V. In vitro antifungal activities of amphotericin B, 5-fluorocytosine, fluconazole and itraconazole against Cryptococcus neoformans isolated from cerebrospinal fluid and blood from patients in Serbia. J. Mycol. Med., 2012, 22(3), 243-248. [http://dx.doi.org/10.1016/j.mycmed.2012.06.002] [PMID: 23518082] |
| [28] | Kerk, G.J.M.; Noltes, J.G.; Luijten, J.G.A. Investigations on organotin compounds. VIII. Preparation of some organotin hydrides. J. Appl. Chem. (Lond.), 1957, 7, 366-369. [http://dx.doi.org/10.1002/jctb.5010070704] |
| [29] | Sheldrick, G.M. SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv., 2015, 71(Pt 1), 3-8. [http://dx.doi.org/10.1107/S2053273314026370] [PMID: 25537383] |
| [30] | Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem., 2015, 71(Pt 1), 3-8. [http://dx.doi.org/10.1107/S2053229614024218] [PMID: 25567568] |
| [31] | Leusink, A.J.; Noltes, J.G. Studies in group IV organometallic chemistry XXXI. Organotin hydride adducts with tin atoms in α,β- or β,β-positions. J. Organomet. Chem., 1969, 16, 91-102. [http://dx.doi.org/10.1016/S0022-328X(00)81639-3] |
| [32] | Mitchell, T.N.; Podestá, J.C.; Ayala, A.; Chopa, A.B. Experimental test of karplus-type relationships for 3J(Sn, C) and 3J(Sn, H). Magn. Reson. Chem., 1988, 26, 497-500. [http://dx.doi.org/10.1002/mrc.1260260612] |
| [33] | Suzuki, I.; Uji, Y.; Kanaya, S.; Ieki, R.; Tsunoi, S.; Shibata, I. Transition-metal-free reductive coupling of 1,3-butadienes with aldehydes catalyzed by dibutyliodotin hydride. Org. Lett., 2017, 19(19), 5392-5394. [http://dx.doi.org/10.1021/acs.orglett.7b02671] [PMID: 28892634] |
| [34] | Ieki, R.; Kani, Y.; Tsunoi, S.; Shibata, I. Transition-metal-free coupling reaction of vinylcyclopropanes with aldehydes catalyzed by tin hydride. Chemistry, 2015, 21(16), 6295-6300. [http://dx.doi.org/10.1002/chem.201406496] [PMID: 25753150] |
| [35] | Podestá, J.C.; Chopa, A.B.; Ayala, A.D.; Koll, L.C. Organotin compounds IX. Reactions between trialkylstannyl-substituted esters and trimethylsilyl halides: Exchange vs. hydrolysis. J. Organomet. Chem., 1987, 333, 25-36. |
| [36] | Thiele, C.M.; Mitchell, T.M. Hydrostannylation of propargylic alcohols using mixed tin hydrides. Eur. J. Org. Chem., 2004, 2, 337-353. [http://dx.doi.org/10.1002/ejoc.200300489] |
| [37] | Podestá, J.C.; Chopa, A.B. Organotin compounds III. Organotin chlorohydride additions to methyl e-disubstituted propenoates. J. Organomet. Chem., 1982, 229, 223-228. [http://dx.doi.org/10.1016/S0022-328X(00)83815-2] |
| [38] | Curran, D.P. Stereochemistry of Radical Reactions: Concepts, Guidelines, and Synthetic Applications, chapter 1., 1996, |
| [39] | CLSI. Reference method for broth dilution antifungal susceptibility testing of yeasts; Approved standard, CLSI Document M27 A3. 2008, |
| [40] | Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother., 2003, 52(1), 1. [http://dx.doi.org/10.1093/jac/dkg301] [PMID: 12805255] |
Endorsements
Browse Contents
Table of Contents
- INTRODUCTION
- MATERIALS AND METHODS
- RESULTS AND DISCUSSION
- ANTIFUNGAL ACTIVITY ASSAYS
- CONCLUSION