- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Arthritis Journal
(Discontinued)
ISSN: 1876-5394 ― Volume 7, 2014
Tolerogenic Dendritic Cells in Clinical Practice
Catharien M.U. Hilkens*, John D. Isaacs*
Abstract
Dendritic cells (DC) play a critical role in maintaining immune tolerance to self-antigens and have become a promising immunotherapeutic tool for treating autoimmune diseases such as rheumatoid arthritis (RA). Tolerogenic DC (tolDC) with stable immunosuppressive function can be generated in the laboratory. These modified tolDC induce antigen-specific T cell tolerance in vitro and in vivo, and can prevent or reduce pathogenic autoimmune responses in experimental animal models of RA. The current challenge is to translate these findings and to develop tolDC for clinical application. In this review we discuss various key considerations for designing tolDC therapy for RA.
Article Information
Identifiers and Pagination:
Year: 2010Volume: 3
First Page: 8
Last Page: 12
Publisher Id: TOARTHJ-3-8
DOI: 10.2174/1876539401003010008
Article History:
Received Date: 29/4/2009Revision Received Date: 11/9/2009
Acceptance Date: 12/9/2009
Electronic publication date: 12/1/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to these authors at the Institute of Cellular Medicine, Musculoskeletal Research Group, Cookson Building, Framlington Place, Newcastle University, Newcastle-upon-Tyne, NE2 4HH, UK; Tel: +44 191 222 8026; Fax: +44 191 222 5455; E-mail: catharien.hilkens@ncl.ac.uk; j.d.isaacs@ncl.ac.uk
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 29-4-2009 |
Original Manuscript | Tolerogenic Dendritic Cells in Clinical Practice | |
INTRODUCTION
Rheumatoid arthritis (RA) is a chronic auto-inflammatory disease that is thought to result from a breakdown in immune regulation. An increased understanding of the role of innate and acquired immunity in RA pathogenesis has led to the development of various immunotherapies. Cytokine neutralisation (TNF, IL-1, IL-6), B-cell depletion and, more recently, T cell co-stimulation blockade with CTLA4-Ig have all been shown to significantly reduce disease activity in RA patients [1, 2]. However, none of these therapies induce a long-lasting, drug-free remission of RA. Moreover, these drugs are likely to compromise protective immunity as they act as general immunosuppressants. It is, therefore, desirable to develop new therapeutic approaches that specifically target the pathologic auto-inflammatory response in RA. A new generation of experimental immunomodulatory therapies is based on the adoptive transfer of autologous regulatory cell populations, such as tolerogenic dendritic cells [3, 4] or regulatory T cells [5, 6]. In autoimmunity these cellular therapies have the potential to restore immune tolerance to self-antigens. Here we discuss the development of dendritic cell (DC)-based therapy for RA.
DENDRITIC CELLS REGULATE IMMUNE TOLERANCE
DC are professional antigen-presenting cells that initiate immune responses. There is compelling evidence that DC not only induce T cell activation, but also instigate peripheral T cell tolerance [7-9]. DC either support or suppress T cell responses. For instance, immature DC that present antigens to T cells in the steady-state will induce tolerance rather than immunity [10-12]. In response to signals associated with infection or inflammation (e.g. pathogen-associated molecular patterns, pro-inflammatory cytokines), immature DC can mature into potent antigen-presenting cells. These immunogenic DC are characterised by the expression of high levels of MHC II, co-stimulatory molecules and pro-inflammatory cytokines and effectively induce effector T cell responses. However, DC maturation does not always lead to immune activation. For instance, DC maturing in response to bacterial LPS in the presence of immunosuppressive agents (e.g. IL-10, TGF-β, glucocorticoids) induce tolerance rather than immunity [13-15].
Mechanisms of tolerance induction by DC include T cell silencing, T cell deletion, polarisation of the T cell cytokine profile (immune deviation) or the induction/expansion of regulatory T cells (Tregs) [7-9]. All these (non-exclusive) mechanisms are believed to be important for the maintenance of peripheral tolerance to self-antigens [16]. A variety of Treg subsets have been described, including naturally occurring CD4+CD25+FoxP3+ T cells which develop in the thymus, and adaptive Tregs that are induced from naïve T cells in the periphery. Examples of induced Tregs are IL-10-producing Tr1 cells, TGF-β-producing Th3 cells and inducible FoxP3+ T cells [16, 17]. DC are central to the induction and control of these various Treg subsets. For instance, immature or semi-mature DC have been shown to induce Tr1 differentiation in vitro and in vivo [10, 18, 19] and DC in the presence of TGF-β induce the development of FoxP3+ Tregs that protect susceptible mice from diabetes [20].
DC AS A THERAPEUTIC TOOL TO INDUCE TOLERANCE
The discovery that DC orchestrate T cell tolerance opened up the possibility that DC could be used as an immunotherapeutic tool for diseases that are characterised by a break-down in immune tolerance [3, 4]. In a proof-of-principle study Dhodapkar et al. demonstrated the feasibility and safety of suppressing influenza-specific effector T cells in humans by the injection of influenza antigen-loaded immature DC in healthy volunteers [21, 22]. The decline in influenza-specific IFN-γ producing T cells in these volunteers lasted for several months and was associated with a rise in IL-10-producing influenza-specific Tregs. These studies are particularly encouraging as they show that it is possible to switch a pre-existing pro-inflammatory effector T cell response into a regulatory T cell response by treatment with immature DC. Thus, it is conceivable that such an approach could be used to modulate the autoimmune response in RA patients and switch it from being auto-aggressive to being auto-suppressive. However, using immature DC for the treatment of RA may be unsafe, as immature DC are not stable: They can lose their tolerogenic state and become immunogenic in response to, for instance, pro-inflammatory cytokines associated with RA (e.g. TNF, IL-1). This would not just counteract their tolerising function, but could also exacerbate the auto-inflammatory response in RA, as immunogenic DC loaded with auto-antigen can induce severe autoimmunity [23]. Similarly, partially or ‘semi’-matured DC have been shown to have tolerogenic function but it was recently demonstrated that these DC are unstable and can become immunogenic in vivo [24, 25]. A preferred option, therefore, is the generation of DC that are in a stable tolerogenic state.
GENERATION OF DC WITH STABLE TOLEROGENIC FUNCTION
There are several strategies to generate stable tolerogenic DC (tolDC) in vitro. DC can be genetically engineered so that they constitutively express immunosuppressive cytokines (e.g. IL-10, IL-4) [26-28], the co-stimulation antagonist CTLA-4 [29, 30] or the apoptosis-inducing Fas-ligand [31]. Another approach is to keep DC in a permanent immature state by inhibiting NFκB, a critical transcription factor for DC maturation. This can be achieved through gene silencing by RNA interference [32, 33], inhibition of NFκB function using decoy oligodeoxyribonucleotides [34, 35] or the synthetic irreversible inhibitor Bay 11-7082 [36], or by downregulation of NFκB components through pharmacological modulation of DC with immunosuppressive drugs [37-39]. For instance, dexamethasone and/or vitamin D receptor agonists (VDR; 1,25(OH)2D3 and its analogues) have been widely used to generate tolDC through the suppression of NFκB-dependent DC maturation [40, 41].
Using mouse models it has been shown that a variety of these in vitro generated tolDC have the ability to inhibit pathogenic auto-reactive T cell responses in vivo. Moreover, tolDC can prevent or even suppress established disease in experimental arthritis models [26, 27, 36, 42-48]. Thus, tolDC show great promise as a cellular tolerogenic tool for the treatment of autoimmune disorders. A first tolDC trial for the treatment of Type I diabetes patients is currently in progress at the University of Pittsburgh (http://clinicaltrials.gov/ ct2/show/study/NCT00445913) and tolDC trials for RA are imminent: We are preparing for a feasibility/safety study with pharmacologically modulated tolDC in RA patients at Newcastle University and another tolDC trial for RA with Bay inhibitor-treated tolDC will commence in the near future at the University of Queensland (Ranjeny Thomas, personal communication).
DEVELOPING tolDC TREATMENT FOR RA
The idea behind tolDC therapy is that it is a highly targeted treatment, only affecting the auto-reactive inflammatory response. It is an autologous therapy where tolDC will be generated from the patient’s own peripheral blood. Modification of DC will take place ex vivo, ensuring that the modifying method (e.g. immunosuppressive drugs) will not affect other immune cells in vivo, hence reducing the risk of general immunosuppression.
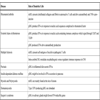
We have developed tolDC for the treatment of RA according to a list of requirements (‘wish list’) as outlined in Table 1. We generate tolDC by pharmacological modulation of monocyte-derived DC with dexamethasone and the VDR agonist 1,25-(OH)2D3, because it is a simple, cheap and effective method, ideal for translation into the clinic. Our tolDC have a typical tolerogenic phenotype with low expression of co-stimulatory molecules and an anti-inflammatory cytokine profile [49]. We found that activation with LPS is necessary to improve the antigen-presenting and migratory capacities of tolDC [50]. Importantly, our tolDC modulate both naïve and memory T cells in a pro-tolerogenic manner in vitro [49, 50], and have a stable tolerogenic phenotype even in the presence of pro-inflammatory cytokines. Furthermore, we have confirmed that these tolDC can be consistently generated from the peripheral blood of RA patients using current good manufacturing practice (cGMP) compatible reagents, such as the safe LPS derivative monophosphoryl lipid A (manuscript in preparation). Although we are satisfied with the in vitro properties of our tolDC, many questions regarding their fate and immunomodulatory effects in vivo remain to be answered. We are currently using a collagen-induced arthritis model to optimise tolDC treatment in terms of dose, route and frequency of tolDC administration, but ultimately these variables can only be definitively addressed by conducting clinical trials in humans.
CHOICE OF AUTO-ANTIGEN
For tolDC to modulate the immune response in an antigen-specific manner they will need to be pulsed with the relevant auto-antigen(s). A key consideration for the design of tolDC therapy for RA patients is, therefore, the choice of auto-antigen. Several auto-antigens have been proposed in RA but auto-reactivity varies between patients and there is no universal auto-antigen to which all patients respond. A pragmatic approach is to use autologous cell-free synovial fluid (SF) as an antigen source. RA SF contains auto-antigens [51, 52], such as type II collagen and human cartilage gp39, and DC can efficiently take up these auto-antigens directly from the SF and process them for presentation to T cells [52]. Thus, SF could be a practical way of loading tolDC with a variety of relevant joint-associated auto-antigens, and could just be simply added to the culture medium during the last stage of in vitro generation of tolDC.
INTRA-ARTICULAR ADMINISTRATION OF tolDC
Another key consideration for the design of tolDC therapy for RA is the route of administration. The most commonly used routes for DC injection (tolDC or mature DC) are intra-venously, -dermally, -lymphatically or -nodally [21, 53-56]. In our proof-of-principle study, we are planning to inject tolDC intra-articularly, into a diseased knee joint. If the anticipated immunoregulatory effects of tolDC spread to other joints, for instance through the induction of regulatory T cells, tolDC injection into a single rather than multiple joints may suffice. tolDC will be administered through an arthroscope, ensuring delivery to the joint cavity whilst also enabling biopsy of the synovial membrane for potential biomarker analysis. This approach will also be suitable for the future treatment of smaller joints.
Administration of tolDC to an inflamed joint has several potential advantages, including i) safety: If the intervention induced unexpected disease exacerbation, it would be possible to treat this locally with, for example, intra-articular corticosteroid; ii) efficacy: Administered tolDC can act locally (the synovial membrane has no basement membrane and contains mature lymphoid follicles in RA [57]) but also systemically, following their anticipated migration to the regional lymph nodes, and iii) biomarkers: Local administration of tolDC provides a unique opportunity to seek biomarkers of effect within the synovial membrane.
CONSIDERATIONS FOR CLINICAL APPLICATION
Whilst cellular therapies appear to be gaining acceptance for the treatment of human disease, a bespoke therapy merits particular consideration. At present our clinical protocol involves harvesting peripheral blood mononuclear cells (PBMC) from a prospective patient and delivering the therapy one week later. It should in the future be possible to cryopreserve tolDC, or to generate them from frozen PBMC or monocytes, but the therapy will remain patient-specific and therefore expensive. This is potentially in contrast to alternative cellular therapies such as mesenchymal stem cells, which may not need to be autologous. Therefore, in its present form, the therapy is likely to be restricted to centres that can both generate and administer the therapy. In this context, a major goal has to be the development of alternative methods to achieve an equivalent product. In particular, the generation of a drug(s) that enables the in vivo targeting of DC or their precursors to trigger differentiation to a tolerogenic phenotype, would be highly desirable. A better understanding of the tolDC surface phenotype and intracellular signalling pathways may assist in such strategies.
Other issues relate to the optimal patient groups for therapy, and its monitoring. The following comments apply equally well to any type of tolerogenic therapy for human autoimmunity. Particularly in a disease such as RA, in which the key immune events probably occur several years before clinical presentation, patients should be considered for tolerogenic therapies as early as possible in their disease course. This presupposes that the treatment has been deemed safe for such a patient group, which may necessitate early trials in more refractory populations. Indeed, with the discovery of anti-CCP, we may even envisage a particularly safe tolerogenic therapy being administered in the ‘pre-clinical’ stage of the disease process. The appropriate monitoring of therapy requires the identification of robust response biomarkers, which is a further research priority. We cannot necessarily assume that a tolerogenic therapy will also be anti-inflammatory and therefore such a therapy may not provide short-term symptom relief when it is first administered. On the other hand, anti-inflammatory drugs could interfere with key mechanisms of tolerance induction and detract from or even oppose the goals of therapy. Of course, inflammation itself may have a similar anti-tolerogenic effect. Under these circumstances biomarkers of tolerance induction may be the only way to recognise that our treatment is working and should continue to be administered. Such surrogate response markers may derive from human or animal studies but are an essential pre-requisite for human therapeutic tolerance trials.
FUTURE PERSPECTIVES
The foregoing should not be taken as a negative view of therapeutic tolerance trials but to merely emphasise the importance of excellent experimental medicine strategies to accompany phase 0, 1 and 2 studies. Increasingly we have the tools to probe deeply into the immune response and, in diseases such as RA, we can do this at the site of disease. Initiatives such as the Immune Tolerance Network (http://www.immunetolerance.org/) have been established with such goals in mind. Given that we can robustly induce tolerance in pre-clinical models there does not seem to be a reason why this could not be translated to the clinic.
In this setting tolDC trials in RA open up a wealth of opportunities for the clinician, the researcher and the patient. Performed in an appropriate setting, and backed up by robust laboratory data and experimental medicine research, we have an opportunity not only to influence the disease course but also to gain a deeper understanding of tolerance mechanisms and biomarkers, to assist the next phase of clinical studies. It is also of prime importance to try to understand the potential influence of anti-rheumatic therapies on tolDC efficacy. For example, some data suggest that anti-TNF drugs could have a parallel influence on DC function in vivo [58, 59]. Furthermore, it could be considered to combine tolDC with other systemic treatments, for instance the VDR agonist 1,25(OH)2D3 (calcitriol), which could potentially reinforce DC tolerogenicity in vivo and at the same time inhibit the progression of bone loss. However, we must also be aware of potentially detrimental interactions between commonly used drugs and tolDC function. For instance it has been reported that COX-2 antagonists can interfere with oral tolerance induction in mouse models [60].
CONCLUSION
In conclusion, tolDC are a promising new immunotherapeutic tool for the treatment of RA and other autoimmune disorders. The therapeutic potential of these immunoregulatory cells has been proven in experimental animal models, and the current challenge is to bring tolDC therapy to the clinic. Although the bespoke nature of this therapy suggests that it will not become a ‘standard of care’ in the near future, lessons learned from tolDC trials are likely to pave the way for the development of new tolerogenic strategies for RA and other autoimmune diseases.
FUNDING
The authors are supported by grants from the Arthritis Research Campaign (arc), Medical Research Council (MRC), Biotechnology and Biological Sciences Research Council (BBSRC) and the J.G.W. Patterson Foundation.
ABBREVIATIONS
| CCP | cyclic citrillunated peptide |
| CCR7 | C-C chemokine Receptor 7 |
| cGMP | current good manufacturing practice |
| CTLA-4 | cytotoxic T lymphocyte antigen-4 |
| DC | dendritic cell(s) |
| FoxP3 | forkhead box P3 |
| IFN | interferon |
| IL | interleukin |
| LPS | lipopolysaccharide |
| MHC | major histocompatibilty complex |
| NFêB | nuclear factor kappa B |
| PBMC | peripheral blood mononuclear cells |
| RA | rheumatoid arthritis |
| TNF | tumour necrosis factor |
| tolDC | tolerogenic dendritic cell(s) |
| SF | synovial fluid |
| TGF-b | transforming growth factor-beta |
| Th3 | T helper type 3 |
| Tr1 | T regulatory type 1 |
| Tregs | regulatory T cells |
| VDR | vitamin D receptor |
REFERENCES
Endorsements
Browse Contents
Table of Contents
- INTRODUCTION
- DENDRITIC CELLS REGULATE IMMUNE TOLERANCE
- DC AS A THERAPEUTIC TOOL TO INDUCE TOLERANCE
- GENERATION OF DC WITH STABLE TOLEROGENIC FUNCTION
- DEVELOPING tolDC TREATMENT FOR RA
- CHOICE OF AUTO-ANTIGEN
- INTRA-ARTICULAR ADMINISTRATION OF tolDC
- CONSIDERATIONS FOR CLINICAL APPLICATION
- FUTURE PERSPECTIVES
- CONCLUSION
- FUNDING