- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Cell Development & Biology Journal
(Discontinued)
ISSN: 1874-0855 ― Volume 3, 2011
Identification of Proteins Interacting with Dysferlin Using the Tandem Affinity Purification Method
Maziar Assadi*, 1, Thomas Schindler1, Bernd Muller1, John D Porter 2, Markus A Ruegg3, Hanno Langen 1
Abstract
Mutations of DYSF, the gene encoding dysferlin, cause two types of muscular dystrophies: limb-girdle muscular dystrophy type 2B and Miyoshi myopathy. Recent work suggests a role of dysferlin in membrane repair and demonstrates that defective membrane repair is a novel mechanism of muscle degeneration. We used the tandem affinity purification method for the purification of proteins interacting with dysferlin. Three interacting partners were identified by this method (striatin, adaptin alpha, utrophin) and were confirmed by co-immunoprecipitations. All three proteins play a role in vesicle trafficking. Knowing the interacting partners of dysferlin will help to understand how muscle cells repair tears in the sarcolemma and will give a deeper insight into this very important cell function. At the same time the identified proteins could serve as potential candidates for other muscular dystrophies and muscle-related diseases with unknown aetiology.
Article Information
Identifiers and Pagination:
Year: 2008Volume: 1
First Page: 17
Last Page: 23
Publisher Id: TOCBJ-1-17
DOI: 10.2174/1874085500801010017
Article History:
Received Date: 31/3/2008Acceptance Date: 18/5/2008
Electronic publication date: 17/6/2008
Collection year: 2008
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the the F. Hoffmann-La Roche, Roche Center for Medical Genomics, Basel, Switzerland; Tel: +41-61-688 25 13; Fax: +41-61-688 14 48; E-mail: maziar.assadi@roche.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 31-3-2008 |
Original Manuscript | Identification of Proteins Interacting with Dysferlin Using the Tandem Affinity Purification Method | |
INTRODUCTION
Dysferlin is a member of a mammalian gene family sharing homology with the Caenorhabditis elegans spermatogenesis factor fer-1 gene, which mediates vesicle fusion to the plasma membrane in spermatids [1].
The human dysferlin gene is located on chromosome 2p13 [2]. Mutations in dysferlin are linked to two clinically autosomal recessive forms of muscle disorders, limb-girdle muscular dystrophy type 2B (LGMD2B) and Miyoshi myopathy (MM) [3,4]. Although the onset of these two diseases is generally in the late teens, they are clinically distinct because they differ in the muscle groups that initially show the onset of the disease. LGMD2B is a predominantly proximal muscular dystrophy. By contrast, MM is a predominantly distal muscular dystrophy. Both diseases progress slowly, and high levels of creatine kinase are detected in the serum of the patients [5,6].
The muscle fibres of dysferlin-null mice maintain a functional dystrophin-glycoprotein complex and a stable structure of the plasma membrane [7,8]. However, these mice develop a progressive muscular dystrophy caused by inefficient resealing of muscle cell plasma membrane [7]. These results suggest that dysferlin has a role in the process of membrane repair in muscle fibres and highlight the importance of this basic cellular function.
Dysferlin is the first identified member of the membrane repair machinery in skeletal muscle. It is highly probable that the protein-binding partners of dysferlin are also members of
this machinery. These proteins could serve as potential candidate for other muscular dystrophies and muscle-diseases with unknown aetiology. Therefore, we attempted to identify the interacting partners of dysferlin and used the tandem affinity purification (TAP) method for this purpose.
MATERIAL AND METHODS
Cloning of Mouse Dysferlin
mRNA was prepared from C57BL/6 mouse skeletal muscle according to the manufacturer’s instruction (Qiagen). cDNA was synthesized with oligo(dT) primer. Dysferlin cDNA was amplified with PCR. The PCR products were inserted into EagI-KpnI site of the vector pBluescript II KS(+) (Stratagene) and transformed into XL1-Blue competent cells (Stratagene). Plasmid DNA was isolated according to the manufacturer’s instruction (Qiagen) and the insert was fully sequence-verified.
Construction of TAP-Tagged Dysferlin
The C-terminal TAP-tagged dysferlin was constructed in two steps. First, the TAP tag was inserted into the SmaI-XhoI site of the vector pSFV. The TAP tag consists of two IgG binding domains of Staphylococcus aureus protein A (ProtA) and a calmodulin binding peptide (CBP) separated by a tobacco etch virus (TEV) protease cleavage site. Second, the entire open reading frame of the mouse dysferlin was inserted into the BssHII-SmaI site of pSFV to generate pSFV-dysferlin-TAP.
Preparation of Recombinant Virus Particles
To produce recombinant Semliki Forest virus (SFV) particles, RNA from in vitro transcribed pSFV-dysferlin-TAP was introduced with RNA from pSFV-helper into BHK cells by electroporation [9]. The electroporated cells were incubated at 31°C. Recombinant virus particles were harvested after 24 h.
Transfection with Recombinant Virus Particles
For checking the activity of the recombinant virus particles mouse myoblast cell line [10] was maintained in 6-well plates and grown in Ham’s F-10 medium (Sigma) supplemented with 15% horse serum (Sigma). The differentiation of myoblasts into myotubes was initiated by reducing the serum content to 2%. 72 h after the induction of myogenesis, cells were infected by adding recombinant virus particles to the cell culture medium. Prior to the transfection, the recombinant SFV particles were activated with chymotrypsin [9]. Cells were harvested 24 h after the transfection and the expression of the tagged dysferlin was checked by Western blot using a peroxidase-antiperoxidase complex (PAP, Sigma) that detects ProtA.
Extract Preparation and TAP Tag Purification
For the TAP tag purification 25 15 cm plates were transfected with recombinant SFV particles as described above. After 24 h cells were scraped, pelleted, lysed in lysis buffer (20 mM HEPES buffer, pH 7.4, 1% Triton X-100) supplemented with protease inhibitor cocktail (Roche) and centrifuged at 10 000 x g for 20 min at 4°C. The supernatant was added to paramagnetic beads coated with a monoclonal human anti-mouse IgG antibody (Dynal Biotech), rotated for 3 h at 4°C, washed 3x with TEV buffer (50 mM Tris, pH 7.4, 100 mM NaCl, 0.1% Tween 20, 1 mM DTT, 5 mM EDTA), resuspended in TEV buffer with TEV protease (Invitrogen) and rotated overnight at 4°C. Eluate obtained after the TEV cleavage was further incubated with calmodulin beads (Stratagene) for 2 h at 4°C. After washing the beads 3x with calmodulin binding buffer (50 mM Tris ph 8.0, 100 mM NaCl, 0.1% Tween 20, 1 mM DTT, 100 mM imidazole, 2 mM CaCl2, 1 mM magnesium acetate), proteins bound to the beads were eluted in the presence of EGTA (25 mM).
Sample Preparation for Mass Spectrometry
The material recovered from TAP tag purification was precipitated with TCA [11], resuspended with LDS sample buffer (Invitrogen) and separated on a 10% SDS-PAGE gel (Invitrogen). After Coomassie staining (Invitrogen), bands were cut out and analyzed by mass spectrometry.
Mass Spectrometric Analysis
The resulting peptides from each gel piece were analyzed by liquid chromatography coupled to a LTQ ion trap mass spectrometer (MS, Thermo Electronics) equipped with a nano-LC electrospray ionization source. Peptides were dissolved in 1% formic acid (buffer A: 0.5% acetic acid, 0.012% heptafluorobutyric acid) and concentrated and desalted online on a C18 PepMap 100 micro precolumn (5 µm particle size, 300 µm x 1 mm; Dionex Corporation) that was coupled to a self packed (7 cm, 3µm; ProntoSil C18-ACE-EPS, Bischoff Chromatography) and pulled (P-2000 laser puller, Sutter Instrument) fused silica capillary (100 μm i.d. × 365 μm o.d.). The chromatographic separation was then performed by a 100 minute nonlinear gradient from 5 to 55% buffer B (80% CAN, 0.5% acetic acid, 0.012% HFBA) with a constant flow rate of 0.2 µl/min. The mass spectrometric data acquisition was performed with a survey scan followed by 7 data dependent MS2 scans with a repeat count of 2. The collision energy was set to 35%. SEQUEST (Thermo Electronics) was used to search the MouseGP database for peptide sequence and protein identification. MouseGP is an in-house protein sequence database that is derived by assembling in sequences the results of Blast searches against the mouse chromosomes of a non-redundant protein set from Swissprot and Trembl. Search parameters included differential mass modification to methionine due to possible oxidation and static mass modification to cysteine due to alkylation by iodoacetamide. Furthermore, one missed cleavage of trypsin was accepted. Peptides identified by SEQUEST may have three different charge states (+1, +2, or +3), each of which results in a unique spectrum for the same peptide. Except in rare instances, an accepted SEQUEST result had to have a ΔCn score of at least 0.1 (regardless of charge state). Peptides with a +1 charge state were accepted if they were fully tryptic or the C-terminus of a protein and had a cross correlation (Xcorr) of at least 1.8. Peptides with a +2 charge state were accepted if they were fully tryptic or partially tryptic between the Xcorr ranges of at least 2.3 and higher. Finally, +3 peptides were only accepted if they were fully or partially tryptic and had an Xcorr > 2.8. Only proteins identified with at least 2 different peptides, with a pepcount (number of identified peptides) larger than 7 were taken into account.
Immunoprecipitaion (IP)
Cells were harvested 96 h after the induction of myogenesis and lysed in the same lysis buffer used for the TAP tag purification. After centrifugation at 10 000 x g for 20 min at 4°C, the supernatant was transferred to a fresh tube and pre-cleared with paramagnetic beads coated with Protein G (Dynal Biotech) for 1 h at 4°C. The beads were separated with a magnet and the supernatant was transferred to a fresh tube. For co-IP mouse monoclonal antibodies were added to the pre-cleared protein extracts: anti-dysferlin (NCL-Hamlet, Novocastra), anti-adaptin alpha (BD Biosciences), anti-striatin (BD Biosciences), anti-utrophin (BD Biosciences). No antibody was added to the control. Samples were incubated overnight at 4°C. After 3 h of incubation with Protein G coupled paramagnetic beads (Dynal Biotech) at 4°C, the protein complex was precipitated with a magnet and the pellet was washed 3x with TEV buffer. Proteins bound to the beads were eluted with LDS sample buffer (Invitrogen) and loaded on a SDS-PAGE gel (Invitrogen). For the Western blots the same antibodies like for the IP were used as primary antibodies and blots were developed with HRP conjugated goat anti-mouse IgG antibody (SouthernBiotech).
RESULTS
To allow purification of proteins associated with dysferlin, we constructed an alphaviral TAP vector. As shown in Fig. (1 ), the TAP cassette, which contains two copies of the IgG binding domains of ProtA of Staphylococcus aureus, the TEV protease cleavage site, and a CBP tag, was fused to the C-terminus of murine dysferlin and was inserted into the pSFV vector.
), the TAP cassette, which contains two copies of the IgG binding domains of ProtA of Staphylococcus aureus, the TEV protease cleavage site, and a CBP tag, was fused to the C-terminus of murine dysferlin and was inserted into the pSFV vector.

To asses whether these tandem affinity tags are in a correct fusion format and to determine the activity of the virus, mouse myoblast cell line was maintained in 6-well plates and transfected with increasing amounts of SFV (10 μl to 500 μl). The expression of the tagged dysferlin in the cell lysates was checked by Western blot. For the detection, we used a complex of PAP which binds to ProtA and allows us to distinguish between endogenous and tagged dysferlin. A specific band of about 230 kDa was visualized in the Western blot (Fig. 2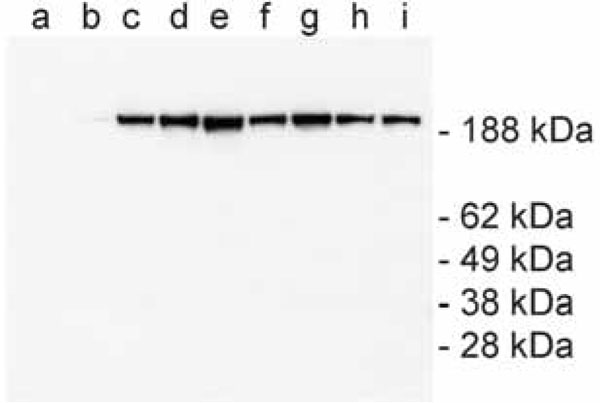 ). The use of higher amount of the virus results at first in an increase and then in a decrease of the expression yield of the tagged dysferlin. This can be explained due to the cytotoxicity of SFV for the host cell. Therefore, the amount in lane e was defined as optimum and was up-scaled for transfection in 15 cm plates.
). The use of higher amount of the virus results at first in an increase and then in a decrease of the expression yield of the tagged dysferlin. This can be explained due to the cytotoxicity of SFV for the host cell. Therefore, the amount in lane e was defined as optimum and was up-scaled for transfection in 15 cm plates.

ProtA binds tightly to IgG-coated beads, which requires the use of TEV protease to elute materials under native conditions. For further purification, the eluate is incubated with calmodulin-coated beads in the presence of calcium. Finally, the complex of dysferlin and its associated proteins is released with EGTA. Fig. (3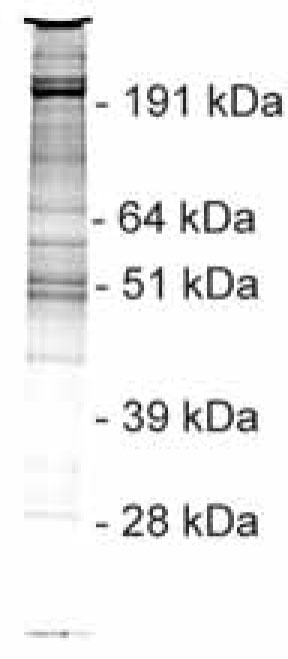 ) shows a Coomassie-stained gel with proteins which were recovered from the purification of TAP-tagged dysferlin.
) shows a Coomassie-stained gel with proteins which were recovered from the purification of TAP-tagged dysferlin.
 |
Fig. (3) TAP purification of dysferlin-TAP tag and its associated proteins. After both purification steps, the proteins were separated using a 10% SDS-PAGE gel and visualized by Coomassie staining. |
Bands shown in Fig. (3) were excised and processed for identification with MS. Table 1 shows the list of the identified proteins with their accession numbers of the UniProt database. Annexin A2, which was previously described as an interacting partner of dysferlin [12], can be found on this list.

A subset of the interacting partners was additionally assessed by co-IP and Western blots. Cell lysates were prepared from our mouse myoblast cell line to determine the interaction of endogenous dysferlin with the following three proteins from the list in Table 1: striatin, adaptin alpha and utrophin. These three proteins were chosen because they are involved either in vesicular trafficking (striatin, adaptin alpha) or muscle regeneration (utrophin). Two different co-IPs were performed for confirming the TAP tag result. First, we used an anti-dysferlin antibody for co-immunoprecipitating the interacting partners of dysferlin. Second, the set-up for co-IP was reversed: we utilised an anti-striatin, -adaptin alpha or -utrophin antibody for co-immunoprecipitating dysferlin. We confirmed by Western blot that either the interacting partners of dysferlin (set-up one, Fig. 4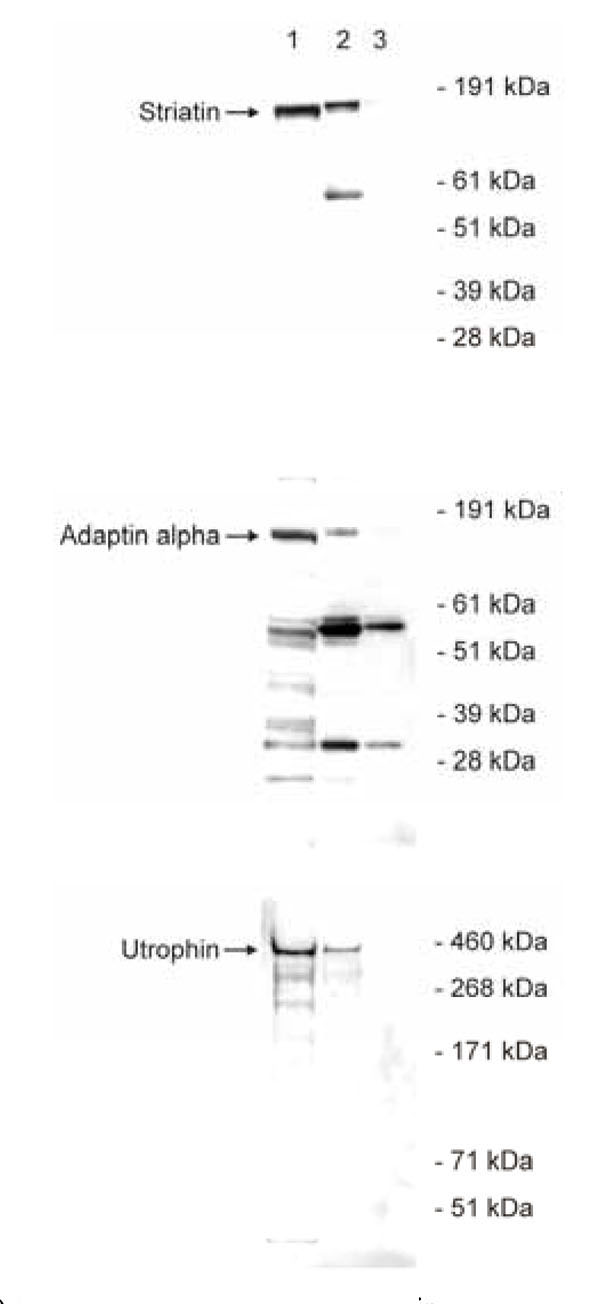 ) or dysferlin (set-up two, Fig. 5
) or dysferlin (set-up two, Fig. 5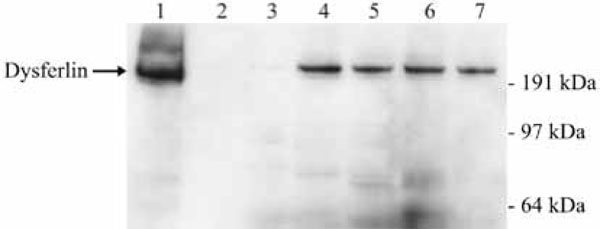 ) were immunoprecipitated. As controls the co-IP procedure was performed in both set-ups without antibodies. The analysis of the co-IPs confirmed that proteins involved in vesicular trafficking or muscle regeneration do indeed interact with dysferlin.
) were immunoprecipitated. As controls the co-IP procedure was performed in both set-ups without antibodies. The analysis of the co-IPs confirmed that proteins involved in vesicular trafficking or muscle regeneration do indeed interact with dysferlin.


DISCUSSION
Disruption of the plasma membrane is a common event in various normal cells. Plasma membrane repair is a basic cellular process required to reseal membrane disruptions [13-15]. The membrane repair machinery allows the cell to overcome physical injuries to the plasma membrane and is essential to prevent disruption-induced cell death. As the primary function of skeletal muscle is to generate physical force, muscle fibres are often exposed to varying degrees of mechanical stress. Therefore, muscle cells are often susceptible to damages of the plasma membrane and require efficient membrane repair machinery.
Dysferlin is a predicted transmembrane protein [8,16]. Mutations in the dysferlin gene cause proximal (LGMD2B) and distal (MM) forms of recessively inherited muscular dystrophies [3,4]. Recent work showed that dysferlin-null mice, like LGMD2B and MM patients, develop a slowly progressive muscular dystrophy [7]. The results of this work suggest a direct role of dysferlin in the repair process of plasma membrane in muscle fibres.
Nearly every major cellular process is carried out by assemblies of large complexes of proteins [17]. It can be assumed that this also pertains for the process of plasma membrane repair. Dysferlin is the first molecule to be discovered that is possibly involved in the membrane repair machinery in skeletal muscle. It is very probable that proteins which interact with dysferlin are associated with this machinery. Furthermore, these proteins are potential candidate genes for other muscular dystrophies. In this work we used the TAP tag purification approach for their identification.
The TAP tag technique allows a spectrometric analysis of dysferlin associated proteins. As usual with such procedures, the interactions must be confirmed by other techniques. This was done by co-IP for the following three proteins: striatin, adaptin alpha and utrophin.
Striatin is an intracellular protein and is involved in ve-sicular trafficking [18]. In the light of recent finding, a dys-ferlin-mediated membrane repair model was already postu-lated [19]. The model states that vesicles are targeted to dis-ruption sites, where they fuse with each other and the plasma membrane. This causes an addition of membrane to the plasma membrane, thereby patching and resealing the dis-rupted membrane. Dysferlin is thought to play a role at the fusion step of this repair process by facilitating the docking and fusion of the vesicles with the plasma membrane through interactions with other proteins.
Adaptins are subunits of adaptor protein (AP) complexes. Four basic AP complexes have been described: AP-1, AP-2, AP-3 and AP-4. Each of these complexes is composed of two large adaptins, one medium adaptin and one small adaptin [20-22]. Adaptin alpha belongs to the group of large adaptins and is part of AP-2. Like striatin, AP complexes are also involved in vesicular trafficking [23].
Early time point of muscle regeneration is characterized by the activation of satellite cells: upon muscle damage quiescent satellite cells start proliferating, migrate to the injury site, and fuse together to repair the damaged fibres [24]. A second type of muscle membrane fusion relates to reseal membrane disruptions which occur under physiological conditions [19]. In both types of membrane fusion, it is thought that vesicles fuse with the plasma membrane to facilitate fusion of apposed membranes [25]. The induction of utrophin during muscle regeneration suggests an important role of it during this process [26-28]. Our TAP data show that dysferlin interacts with utrophin. This result was also confirmed by IP. Therefore, we propose that there is a link between muscle regeneration and the repair of muscle membrane and that dysferlin plays an important role during both processes. Inefficient regeneration of muscle fibres contribute to reduced muscle mass seen with age. Thus, dysferlin and its associated proteins could not only be important for disease states, but also for muscle degeneration associated with aging.