- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Cell Development & Biology Journal
(Discontinued)
ISSN: 1874-0855 ― Volume 3, 2011
Nitric Oxide and Cellular Maturity Are Key Components of Pro-Inflammatory Cytokine-Induced Apoptosis of Human Fetal Lung Epithelial Cells
Michael A Posencheg1, Andrew J Gow2, Ping Wang1, Linda W Gonzales1, Changjiang Guo*, 2
Abstract
Inflammation is a major contributor to the pathogenesis of bronchopulmonary dysplasia (BPD). BPD is associated with prematurity of birth, sepsis, with increased production of both cytokines and nitric oxide, and with the shedding of bronchial epithelial cells. The pathological mechanisms involved in this disease remain unclear, in particular the role that epithelial maturity plays. The effects of pro-inflammatory cytokines upon immature and mature cells are examined within this study, using primary culture of human lung epithelial cells. Pro-inflammatory cytokines increase inducible nitric oxide synthase (iNOS) expression and raise NO production, irrespective of cellular maturity. Preincubation with 1400W, a specific iNOS inhibitor, abrogated pro-inflammatory cytokine-induced NO generation and apoptosis. However, immature fetal lung epithelial cells were uniquely sensitive to cellular injury in response to cytokine exposure. These observations suggest that pro-inflammatory cytokines, which are present within BPD, may cause apoptosis of lung epithelial cells via de novo generation of NO. Furthermore, the prematurity of lung epithelial cells may be a factor in free radical mediated pulmonary damage.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 3
First Page: 1
Last Page: 5
Publisher Id: TOCBJ-3-1
DOI: 10.2174/1874085501103010001
Article History:
Received Date: 31/7/2010Revision Received Date: 20/12/2010
Acceptance Date: 13/1/2011
Electronic publication date: 15/04/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at Rutgers, The State University of New Jersey, Department of Pharmacology & Toxicology, 160 Frelinghuysen Road, Piscataway, NJ 08854, USA; Tel: (732) 445-6190; Fax: (732) 445-1191; E-mail: guoc@rci.rutgers.edu
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 31-7-2010 |
Original Manuscript | Nitric Oxide and Cellular Maturity Are Key Components of Pro-Inflammatory Cytokine-Induced Apoptosis of Human Fetal Lung Epithelial Cells | |
INTRODUCTION
Bronchopulmonary dysplasia (BPD), which occurs in the lungs of premature infants, is defined as a requirement for supplemental oxygen at 36 weeks postmenstrual age [1]. This pathophysiology appears to be caused by a combination of factors including inflammatory injury, mechanical ventilation, oxygen therapy and lung prematurity. When first described by Northway in 1967, premature infants who developed the disease were more mature (31-34 weeks gestation) than those seen today [2]. Because of advances in mechanical ventilation, surfactant replacement therapy and improved neonatal intensive care, the incidence of BPD in infants older than 30 weeks gestation has decreased. However, in more premature infants (gestational age less than 28 weeks) and those of a very low birth weight (less than 1,500g) BPD continues to be problematic [3-5]. Infants described by Northway typically had a history of severe hyaline membrane disease, whereas today, the disease appears to be manifested primarily by arrested alveolar development [6]. While treatment induced oxygen injury and volutrauma continue to play a role in BPD, extreme lung immaturity, inflammation and multiple other factors have also been implicated in animal studies [7]. Nitric Oxide, which is produced endogenously by the three isoforms of the enzyme nitric oxide synthase (NOS) (neuronal, inducible and endothelial), plays an important role within the functioning airway epithelium [8-10]. As well as being critical to normal lung physiology, NO is also recognized to play a key role in several inflammatory airway diseases, including BPD. Specifically it has been shown that hyperoxia, a causative factor in BPD, results in a loss of NO-mediated airway relaxation. In a fetal baboon model of BPD, a decreased level of two of the three NOS isoforms (neuronal and endothelial) and a parallel down regulation of NO production was found to be characteristic in the early postnatal period of the disease [11].
In the lung, epithelial cells play a critical role in host defense, as they form the first line of interaction with the external environment. Due to the specialized nature of the epithelial cells involved in forming this protective barrier, their maturation is critical to organ systems such as the lung and the intestine. Incomplete maturation of the epithelial lining may provide a key to the origin of many prematurity-associated diseases, such as BPD. Cytokines, and their induction through inflammation and/or infection, are a critical factor in epithelial injury, especially in neonatal disease [12]. Pulmonary inflammation is often associated with increased expression of iNOS. First identified in macrophages [13], iNOS is expressed by many different cell types [14]. Excessive NO via iNOS is associated with a number of inflammatory diseases and previously we have shown that increased iNOS expression and NO-mediated modification is associated with BPD [15].
This study is based on the hypothesis that the maturity of pulmonary epithelial cells is a critical factor in determining the inflammatory response induced by cytokines. In order to test this hypothesis, we have utilized a unique in vitro primary cell culture system. In this system, human fetal lung alveolar epithelial cells are used to generate a maturity-based model. Culturing the cells in serum-free Waymouth’s medium allows them to retain the undifferentiated epithelial phenotype, typically found in the developing neonate. Addition of dexamethasone, cAMP and isobutylmethylxan-thine (DCI), to the medium induces the cells to take on a functionally mature type II cell phenotype, as revealed by changes in gene expression and cellular function [16]. This model provides a unique opportunity to study mature type II epithelial cells (mature fetal lung epithelial cell, mFLEC) in comparison to the undifferentiated immature epithelial cells found in the developing human neonate (immature fetal lung epithelial cell, iFLEC). Within this study we have examined the response of these two cell types to pro-inflammatory cytokine exposure.
MATERIALS AND METHODS
Cell Culture
Human fetal lung was obtained from second trimester therapeutic abortions (13-20 week gestation) under protocols approved by the Committee for Human Research, Children’s Hospital of Philadelphia. The fetal lung parenchyma was dissected free of large airways, chopped into 1mm3 explants, and cells isolated by enzymatic digestion as described [16]. These cells, once isolated and purified, were cultured for 5 days on 35-mm dishes in serum-free Waymouth’s medium alone (immature fetal lung epithelial cell, iFLEC) or containing a mixture of Dexamethasone (10 nM), cAMP (0.1mM), and isobutylmethylxanthine (0.1 mM) (mature fetal lung epithelial cell, mFLEC) as described previously [16]. Cells were maintained in a sterile incubator at 37(C and 5% CO2 throughout the experiments. Pro-inflammatory cytokine exposure was achieved by exposing both cell types to the cytokines IFN-( (2,500U/ml) and IL-1( (10,000U/ml) and examining responses over time.
Cell Death Determination
YoPro-1 (Molecular Probes, Carlsbad, CA), 0.5 µM, was added to the medium during cytokine exposure. Rhodamine 123 (Molecular Probes), 10 μM, was added prior to cytokine exposure. Cells were examined utilizing an inverted fluorescence microscope (Nikon, Melville, NY) and a Metamorph imaging software (Universal Imaging Corp., PA). Live and dead cells were counted in 3 fields per dish.
Preparation of Cell Lysate
Cells were harvested on ice. The media was removed and stored at -80(C, and the cells were washed 3x in cold PBS. 100 µl of lysis buffer (Hepes 20 mM, NaCl 150 mM, Glycerol 10%, Triton X-100 1%, EGTA 1mM, MgCl2 1.5mM, pH 7.4) containing protease inhibitors (PMSF 1mM, NaPyrophosphate 10mM, NaF 50 mM, Na Orthovanadate 2mM, Lactacystin 1μM, AEBSF 1mM, EDTA 0.5mM, Bestatin 65 µM, E-64 0.7 μM, Leupeptin 0.5µM, and Aprotinin 0.15 μM) was added and the cells were scraped. Cell lysate solution was sonicated at 5 Watts for 10 seconds and stored at -80(C.
Western Blot Analysis
40 μg of cell lysate protein (as determined by Bradford assay) was electrophoresed on a 4-12% 1D SDS-PAGE Gel (Invitrogen, Carlsbad, CA). Samples were transferred to a PVDF membrane at 20 volts overnight using a tris/glycine/20% methanol transfer buffer. Blots were examined as described previously [17] using a mouse monoclonal iNOS primary antibody at 1:500 dilution (Becton-Dickinson, Franklin Lakes, NJ) and goat anti-mouse IgG (H+L)-HRP conjugate secondary antibody at 1:5000 dilution (Bio-Rad, Hercules, CA). Blots were visualized by ECL detection kit as per manufacturer’s instructions (Amersham, Piscataway, NJ).
Nitrate and Nitrite Analysis
A Nitrate/Nitrite Colorimetric Assay Kit (Cayman Chemical, Ann Arbor, MI) was utilized for analysis of total nitrogen oxides in the cell culture media. Media were analyzed as per manufacturer’s instructions using a two-step method involving nitrate reduction and nitrite detection via a modified Griess assay.
Caspase-3 Activity
Caspase-3 activity within cell lysates was measured using a Fluorometric Assay Kit (Sigma, St. Louis, MO), without the addition of cysteine protease inhibitors in the lysis buffer solution. Activity is assessed by release of 7-amino-4-methycoumarin from the substrate Ac-DEVD-AMC. Fluorescence was measured using 360 and 460 nm as the respective excitation and emission wavelengths. Results are presented as caspase-3 activity/μg of protein.
Electron Microscopy
Cells were processed and analyzed, after fixing in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1M Sodium Cacodylate overnight at 4(C. The resulting cell pellet was rinsed in 0.1M Sodium Cacodylate then postfixed with 2% osmium tetroxide, dehydrated in graded ethanol, and embedded in Epon. Sections (70 nm) were examined with the JEOL JEM 1010 electron microscope after uranyl acetate and bismuth subnitrite staining. Images were captured using a Hamamatsu ccd camera and AMT 12-HR software.
Statistical Analysis
Data with multiple groups were analyzed by ANOVA based on the number of dependent variables using SigmaStat software.Pairwise analysis using a Tukey test or one-sided t-test was performed in the setting of a significant ANOVA, using p<0.05 as a significant result. Values are presented as mean ± standard deviation.
RESULTS
Pro-Inflammatory Cytokines Induce Apoptosis of Human Fetal Lung Epithelial Cells
After incubation with or without cytokines for 24 h,electron microscopic examination of immature and mature human fetal lung epithelial cells is shown in Fig. (1A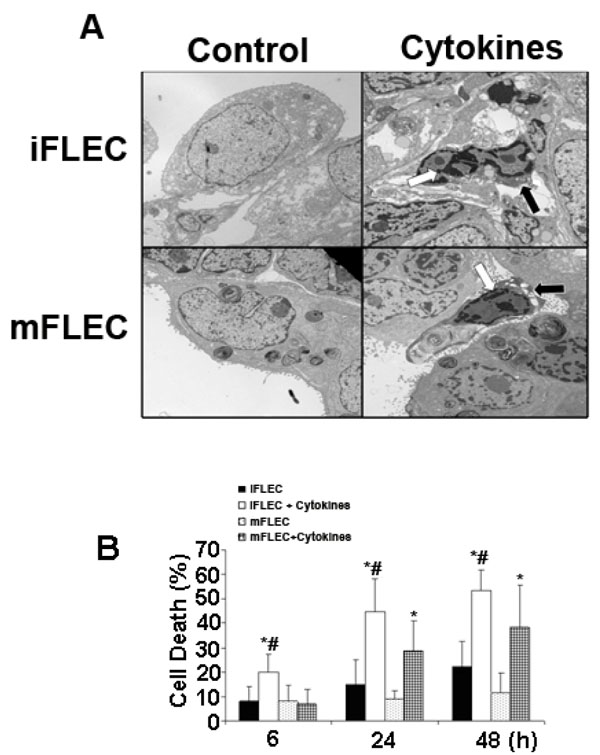 ). The formation of large multivesicular organelles, termed lamellar bodies, is a striking characteristic of mature Type II pneumocytes. At baseline, mFLEC can be seen to contain mature lamellar bodies that are less apparent in iFLEC. These observations confirm DCI treatment as an effective methodology for eliciting cellular maturation, prior to cytokine stimulation. With cytokine exposure, various degrees of nuclear and cytoplasmic condensation are observed in both mFLEC and iFLEC. These condensations are consistent with cytokine-mediated induction of apoptosis in both cell types.
). The formation of large multivesicular organelles, termed lamellar bodies, is a striking characteristic of mature Type II pneumocytes. At baseline, mFLEC can be seen to contain mature lamellar bodies that are less apparent in iFLEC. These observations confirm DCI treatment as an effective methodology for eliciting cellular maturation, prior to cytokine stimulation. With cytokine exposure, various degrees of nuclear and cytoplasmic condensation are observed in both mFLEC and iFLEC. These condensations are consistent with cytokine-mediated induction of apoptosis in both cell types.

In order to gauge the degree of cell death induction, cells were examined over time by the use of Yo-Pro and R123 labeling [18]. In iFLEC, cytokine-treatment induced significant apoptosis at 6, 24 and 48 hours. At 6 hours, cytokine exposure resulted in 19.9 ± 7.3% cell death vs 8.1 ± 6.1% in controls (p<0.05). After 24 hours, cell death was 44.6 ± 13.6% in cytokine exposed cells vs 14.7 ± 10.3% in controls (p<0.05). Finally, 48 hours of cytokine exposure produced 53.4 ± 8.6% cell death vs 22.1 ± 10.7% in the control group (p<0.05) (Fig. 1B).
mFLEC also exhibited a statistically significant increase in apoptosis when exposed to pro-inflammatory cytokines but the time course of cell death differed from that observed in iFLEC. mFLEC showed no increase in apoptosis measured at 6 hours (6.7 ± 6.1% vs 8.1 ± 6.4%, NS). However, at both 24 and 48 hours, there was a statistically significant increase in cell death (24 hours: 28.6 ± 12.3% vs 8.8 ± 3.4%, p<0.05; 48 hours: 38.1 ± 17.7% vs 11.4 ± 8.2%, p<0.05) (Fig. 1B). As well as exhibiting a delayed cell death response mFLEC displayed a reduced sensitivity to cytokine-mediated apoptosis. Cell death related to cytokine exposure in iFLEC was significantly greater than that in mFLEC at all time points (6 hours: 19.9 ± 7.3% vs 6.7 ± 6.1%, p<0.05; 24 hours: 44.6 ± 13.6% vs 28.6 ± 12.3%, p<0.05; 48 hours: 53.4 ± 5.6% vs 38.1 ± 17.7%, p<0.05). (Fig. 1B).
Role of Cellular Maturity in Cytokine-Induced Activation of Caspase-3
Caspase-3 activation, which is an effector caspase within the apoptotic pathway, occurs upstream of DNA damage in the apoptotic pathway. Caspase-3 activity was significantly increased in iFLEC with cytokine exposure at all time points (Fig. 2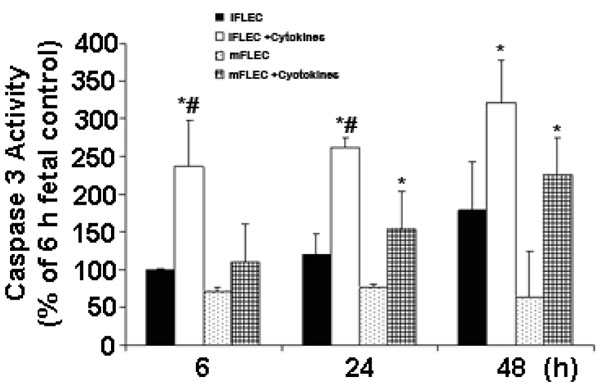 ). In mFLEC, caspase-3 activity was not altered at 6 hours, but was increased at both 24 and 48 hours in cytokine-exposed vs unexposed cells (Fig. 2). When caspase-3 activation was compared between cytokine-exposed iFLEC and mFLEC, a significant increase within iFLEC was observed at both 6 and 24 hours (6 hours: 236 ± 61.4 vs 110 ± 20.4, p<0.05; 24 hours: 261 ± 14.8 vs 153 ± 8.3, p<0.05, expressed as % of 6 hour iFLEC control [=100] and the basal level of caspase 3 was 73±15 (RFU-relative fluorescence unit, mean ± SD, n=6) of iFLEC at 6h. This increase in caspase-3 activation in immature cells at earlier time points than mature human fetal lung epithelial cells is consistent with an earlier induction of the cell death process in immature human fetal lung epithelial cells.
). In mFLEC, caspase-3 activity was not altered at 6 hours, but was increased at both 24 and 48 hours in cytokine-exposed vs unexposed cells (Fig. 2). When caspase-3 activation was compared between cytokine-exposed iFLEC and mFLEC, a significant increase within iFLEC was observed at both 6 and 24 hours (6 hours: 236 ± 61.4 vs 110 ± 20.4, p<0.05; 24 hours: 261 ± 14.8 vs 153 ± 8.3, p<0.05, expressed as % of 6 hour iFLEC control [=100] and the basal level of caspase 3 was 73±15 (RFU-relative fluorescence unit, mean ± SD, n=6) of iFLEC at 6h. This increase in caspase-3 activation in immature cells at earlier time points than mature human fetal lung epithelial cells is consistent with an earlier induction of the cell death process in immature human fetal lung epithelial cells.

Pro-Inflammatory Cytokine–Induced Apoptosis of Human Fetal Lung Epithelial Cells is NO Dependent
NO is produced upon activation by pro-inflammatory cytokine exposure in a wide spectrum of cell types, including pulmonary epithelial cells and alveolar macrophages [19], and has been implicated in the induction of cell death pathways. Incubation of both iFLEC and mFLEC with pro-inflammatory cytokines increased expression of iNOS and resulted in de novo generation of NO, as shown by nitrite release to the medium (Fig. 3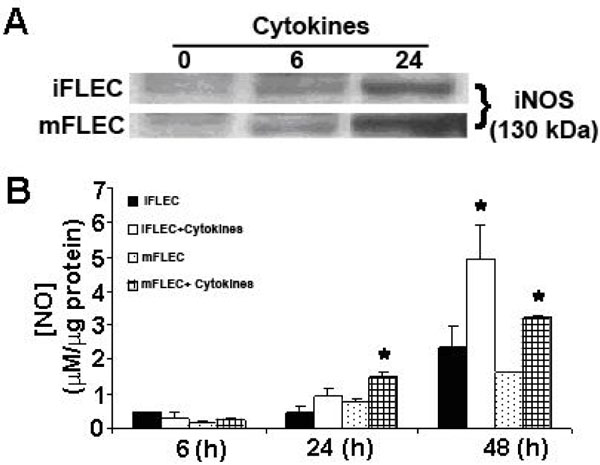 ). In order to determine whether nitric oxide is a factor in cytokine-induced cell death, cells were pre-incubated with the specific iNOS inhibitor 1400W prior to 24 h of cytokine treatment. 1400W significantly reduced cytokine-mediated increases in NO production in both iFLEC and mFLEC (Fig. 4A
). In order to determine whether nitric oxide is a factor in cytokine-induced cell death, cells were pre-incubated with the specific iNOS inhibitor 1400W prior to 24 h of cytokine treatment. 1400W significantly reduced cytokine-mediated increases in NO production in both iFLEC and mFLEC (Fig. 4A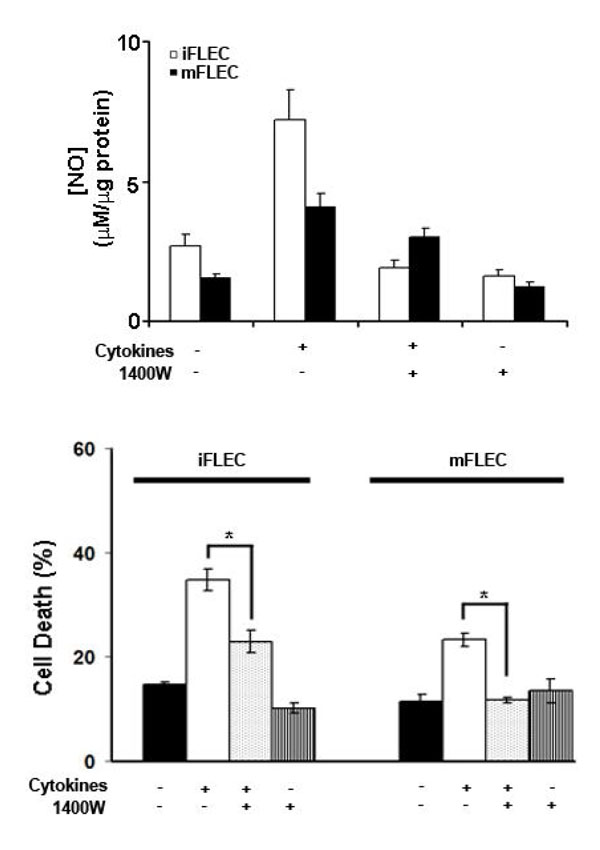 ). Importantly, 1400W did not completely inhibit nitrite release from these cells, as there is significant expression of the nNOS isoform within pulmonary epithelial cells. Pre-treatment of both iFLEC and mFLEC with 1400W significantly reduced cytokine-mediated cell death, in accordance with iNOS-derived NO playing a significant role in the induction of apoptosis (Fig. 4B).
). Importantly, 1400W did not completely inhibit nitrite release from these cells, as there is significant expression of the nNOS isoform within pulmonary epithelial cells. Pre-treatment of both iFLEC and mFLEC with 1400W significantly reduced cytokine-mediated cell death, in accordance with iNOS-derived NO playing a significant role in the induction of apoptosis (Fig. 4B).


DISCUSSION
BPD is a chronic lung disease associated with premature birth and characterized by early lung injury and associated with increased expression of pro-inflammatory cytokines [20]. Cytokines play key roles in the regulation of the immune response to inflammatory stimuli such as tissue injury. Although there have been numerous studies that have examined the role that cytokines play in regulating epithelial cells, little is known regarding the effects of cellular maturity upon these responses. This study utilized a unique cell culture system, which models both the mature and the immature pulmonary epithelium to investigate the responsiveness of these cells to inflammatory signaling. Herein we have demonstrated that in both cell types the pro-inflammatory cytokines IFN-( and IL-1( induce apoptotic cell death in a manner that is in part dependent upon iNOS-derived NO. Furthermore, it appears that immature epithelial cells have an accentuated response implying that the maturity of the epithelial cell may be a critical factor in the development of inflammatory lung disease, such as BPD.
Previously, we have shown that primary culture of human fetal lung epithelial cells can be used as a maturity-based epithelial cell model [16]. Undifferentiated epithelial cells incubated in Waymouth’s medium alone do not produce surfactant components and thus retain a relatively undifferentiated state akin to the fetal epithelium. However, culture for 4 days in a mixture of dexamethasone, cAMP and isobutylmethylxanthine results in a differentiated mature type II cell phenotype with normal surfactant processing [16]. Thus, the control epithelial cells are a reasonable model for an immature form of epithelial cell that would be present in a severely premature infant, while the DCI-treated cells are more akin to the mature surfactant-producing type II alveolar epithelial cells of a term infant.
Consistent with earlier studies of both human and rat fetal lung cells, pro-inflammatory cytokines induced apoptosis in both mature and immature human lung epithelial cells. Since the typical pathology of BPD is associated with premature birth, we hypothesized that cellular maturity may play a key role in pro-inflammatory cytokine-mediated apoptosis. Our data showed that the immature cells, iFLEC, were more sensitive to pro-inflammatory cytokine induced cell death than the mature cells, mFLEC. Within these cells cytokine-mediated cell death appears to be predominantly apoptotic as indicated by electronic microscopy analysis and caspase-3 activation. In both iFLEC and mFLEC, functional expression of iNOS in response to cytokines resulted in an increase in total nitrogen oxide production. Elevated levels of reactive oxygen and nitrogen species (RONS) have been detected in patients with forms of acute lung injury (ALI) and ARDS in bronchial-alveolar lavage fluid and pulmonary edema fluid, correlating with severity of injury to the alveolar epithelium [21]. Recently, we have demonstrated alteration in NOS expression and production of NO metabolites in lung tissue from infants with end stage BPD [15]. It has also been demonstrated in rat type II cells that the addition of NO results in decreased ATP and surfactant synthesis [22]. That iNOS-derived NO is critical to the cell death response is demonstrated by the specific inhibitor 1400W, which reduces total NO production and cell death following cytokine-treatment (Fig. 4). Although the role of NO in cell death remains unclear, it is generally thought that within cytokine responsive cells, such as macrophages, NO production is critical to their anti-infection role and induces consequent apoptosis. It would appear that within pulmonary epithelial cells this pattern is mirrored, indicating that these cells may play an active role in resistance to infection, rather than being a mere barrier.
Although our in vitro observations cannot be extrapolated directly to the in vivo situation, pro-inflammatory cytokine-induced apoptosis of fatal lung epithelial cells may have pathophysiological relevance. Recent studies suggest that both pro-inflammatory cytokines and NO are factors that affect disease severity of premature related diseases such as NEC and BPD. Human lung epithelial cells are responsive to inflammatory stimuli, such as cytokine exposure, and the relative maturity of lung epithelial cells influences the degree to which they respond. The sensitive nature of fetal cells to cytokines may provide insight to understanding the pathogenesis of inflammatory diseases in premature infants. A better understanding of cellular maturity based signaling events will enable design of therapeutic approach by a targeted strategy in order to control inflammatory diseases in premature infants, such as BPD.
CONFLICT INTEREST
The authors have declared that no conflicting interests exist.
ACKNOWLEDGEMENTS
The authors thank Ravi Savani, Trevor Fusaro and Pamela Scott for assisting in this study. This study was partially supported by grant from the National Institutes of Health, HL 074115 (AJG).