- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Condensed Matter Physics Journal
(Discontinued)
ISSN: 1874-1924 ― Volume 4, 2013
First Principles Investigation of Ferromagnetism for Zn1-xMnxY(Y = S, Se, Te)
N Benkhettou*, D Bensaid
Abstract
Through first-principles full-potential linear muffin-tin orbital (FP-LMTO) method within the local density approximation, we investigate the structural and electronic properties of Zn1-xMnx Y (VIa) where Y represent an element of the VIa column(S, Se and Te) compounds-based diluted magnetic semiconductors (DMS) in the Zinc-blend structure. We have investigated the lattice parameters and band gap energies. The lattice constants a are found to change linearly for the Zn1-xMnxTe and Zn1-xMnxS alloy
Article Information
Identifiers and Pagination:
Year: 2008Volume: 1
First Page: 29
Last Page: 34
Publisher Id: TOCMPJ-1-29
DOI: 10.2174/1874186X00801010029
Article History:
Received Date: 28/9/2008Revision Received Date: 10/11/2008
Acceptance Date: 11/11/2008
Electronic publication date: 18/12/2008
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License(http://creativecommons.org/licenses/by-nc/2.5/), which permits unrestrictive use, distribution, and reproduction in any medium, provided the original work is properly cited.
* Address correspondence to this author at the Department of Physics, Faculty of sciences, University of Sidi Bel Abbes, Algeria; Tel/Fax: (213) 48 54 43 44; E-mails: nordine_bt@yahoo.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 28-9-2008 |
Original Manuscript | First Principles Investigation of Ferromagnetism for Zn1-xMnxY(Y = S, Se, Te) | |
INTRODUCTION
Recently, diluted magnetic semiconductors (DMS) have attracted the attention of the academic and industrial communities, since they can be used in developing spintronics, integrated optoelectronic devices, and nano-structured quantum devices, because spin-related phenomena as well as carrier transport can be manipulated in a well-controlled semiconductor system [1-4]. II-VI compounds show in addition novel magneto-optical and magneto-transport properties if the cation is partly substituted by Mn with its half-filled 3d shell. The interaction between Mn 3d electron and the electronic states of the host crystal is of special interest [5].
The variation in the relative intensities of the Mn 3d features by changing the anion in a compounds series, like Cd1-xMnxY or Zn1-xMnxY (Y = S, Se, Te), is promising to clarify the amount of p-d hybridization in these alloys.
In this paper, we investigate the magnetism of ZnS, ZnSe and ZnTe based DMSs based on ab initio electronic structure calculations in order to have clues about the origin of the ferromagnetism in DMSs [6-8]. In a II–VI compound, transition metal (TM) impurities do not introduce any carriers, therefore, we can discuss effects of carrier doping separately from a concentration of TM impurities. In addition to that, detailed investigation on the feasibility of II–VI compound-based DMS is of great importance from the industrial view point, because it is easy to dope TM impurities into II–VI compounds up to several ten percent [9].
Calculation
The electronic structure calculations of the DMS are performed based on the local density approximation with the parameterization by Perdew-Wang [10]. For the (FP-LMTO) calculations, we have used the available computer code Lmtart [11].
We employ the muffin-tin approximation to describe the crystal potential, and the wave functions in each Muffin-tin sphere are expanded with real spherical harmonics up to lmax = 6. The k integration over Brillouin zone is performed using the tetrahedron method [12].
ZnS, ZnSe, and ZnTe have the zinc blende crystal structure, and their lattice constants are a = 5.4093 A°, a = 5.6676 A ° and a = 6.089 A°, respectively. Muffin-tin radii are chosen so that they touch with each other
RESULTS AND DISCUSSION
Structural Properties
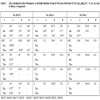
We first calculated the structural properties of the binary compounds ZnSe, ZnTe, ZnS and MnTe, MnSe, MnS in the zincblende structure. As for the semiconductor ternary alloy in the type B1-xAxC, we have started our FP-LMTO calculation of the structural properties with the zinc-blende structure and let the calculation forces to move the atoms to their equilibrium positions. For divers concentrations x = 0.25, 0.5, 0.75. We perform the structural optimization by calculating the total energies for different volumes around the equilibrium cell volume V0 of the binary and their alloy. The calculated total energies are fitted to the Murnaghan’s equation of state [13] to determine the ground state properties such as the equilibrium lattice constant a0, the bulk modulus B and its pressure derivative B’. The calculated equilibrium parameters (a0, B and B’) are given in Table 1 Our results are compared with the experiment and with the FP-LAPW (method) results.
Usually, in the treatment of alloys, it is assumed that the atoms are located at the ideal lattice sites and the lattice constant varies linearly with the composition x according to the so-called Vegard’s law [14]:
a (AxB1-xC) = xaAC + (1 – x) aBC, (1)
where aAC and aBC are the equilibrium lattice constants of the binary compounds AC and BC, respectively, and a (AxB1-xC) is the alloy lattice constant. The obtained equilibrium lattice constant, of the ferromagnetic Zn1-xMnxY (Y = S, Se, Te) semi-magnetic semiconductors are given in Table 1.
The equilibrium lattice parameter of the zinc-blende Zn1-xMnxTe and Zn1-xMnxS. We found a good agreement with the experiment on the other. We have determined the equilibrium lattice parameter of the zinc-blende MnY (Y = S, Se, Te), which is found to be underestimated compared to the experimental.
The equilibrium lattice constants a shows a linear variation versus Mn concentration of Zn1-xMnxS and Zn1-xMnxTe, but a small deviation from Vegard’s law is clearly visible for the Zn1-xMnxSe.
There are no results to compare with our values. Thus finds new values for various concentrations, our result to take as reference. Fig. (1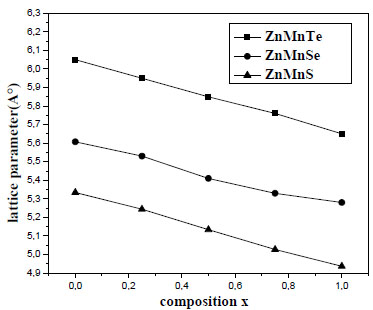 ), show the variation of the calculated equilibrium lattice constant versus concentration x for Zn1-xMnxY(Y = S, Se, Te). The Mn mole fraction (x) was determined by using the linear relation: a(x) = -0.4044X + 5.3374 (A°).
), show the variation of the calculated equilibrium lattice constant versus concentration x for Zn1-xMnxY(Y = S, Se, Te). The Mn mole fraction (x) was determined by using the linear relation: a(x) = -0.4044X + 5.3374 (A°).
 |
Fig. (1) Variation of the calculated equilibrium lattice constant versus concentration x for Zn1-xMnx Y (Y = S, Se, Te). |
Obtained for lattice parameter of Zn1-xMnxS and lattice parameter for Zn1-xMnxSe vary according to the linear relation: a(x) = - 0.3416X + 5.6022 (A°).
Fig. (2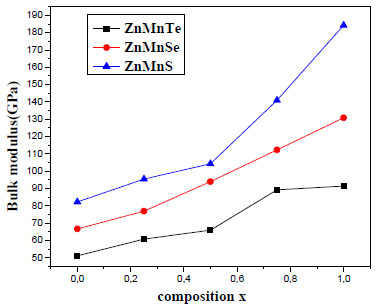 ) shows the variation of the bulk modulus versus concentration. The bulk modulus grew linearly with the composition Mn for the Zn1-xMnxSe.
) shows the variation of the bulk modulus versus concentration. The bulk modulus grew linearly with the composition Mn for the Zn1-xMnxSe.
 |
Fig. (2) Variation of the calculated bulk modulus versus concentration x for Zn1-xMnxY(Y = S, Se, Te). |
We observe in Fig. (2) that bulk modulus grew with concentration (x) and with the nuclear charge of the element of the VIa column.
Electronic Properties
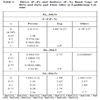
The calculated band structure energies of binary compounds as well as their alloy using FP-LMTO method within LDA approximation exhibit a direct band gap at Г point for Zn1-xMnxY(Y = Zn, Se, S)with x vary between 0.25 and 0.75, the valence band maximum (VBM) and the conduction band minimum (CBM) occur at the Г and X, respectively. The important features of the band structure (main band gaps) are given in Table 2.
As well as the theoretical and experimental ones. It is clearly seen that the band gap are underestimated in comparison with experiment results. This underestimation of the band gaps is mainly due to the fact that the simple form of LDA does not take into account the quasiparticle self-energy correctly [25].
The carrier induced ferromagnetism by hole doping was observed in (Zn, Mn) Te [32], and II–VI based-DMSs have been of much interest also from the scientific point of view. Taking these aspects into account, it is important to investigate the carrier induced effects in ZnS, ZnSe and ZnTe based DMSs.
Electron Band Structures and Densities of States
In order to elucidate underlying mechanism of the ferromagnetism in II–VI compound-based DMSs, the density of states (DOS) is calculated. Figs. (3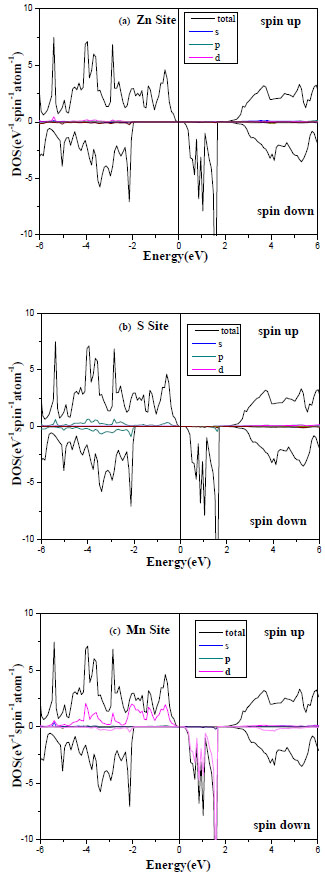 -5
-5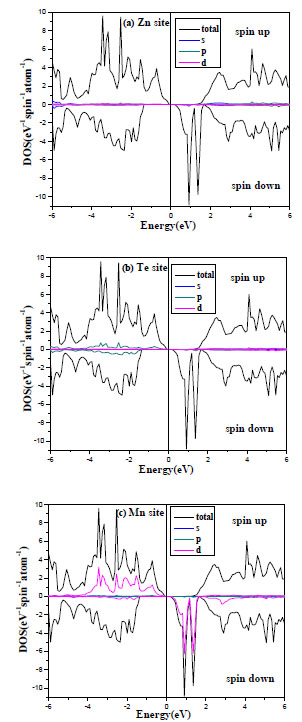 ) shows the DOS of 25% TM-doped ZnTe based DMSs in the ferromagnetic state. It is found that the impurity states appear in the band gap.
) shows the DOS of 25% TM-doped ZnTe based DMSs in the ferromagnetic state. It is found that the impurity states appear in the band gap.
 |
Fig. (3) DOS for the ferromagnetic Zn1-xMnxS for spin up and spin down. |
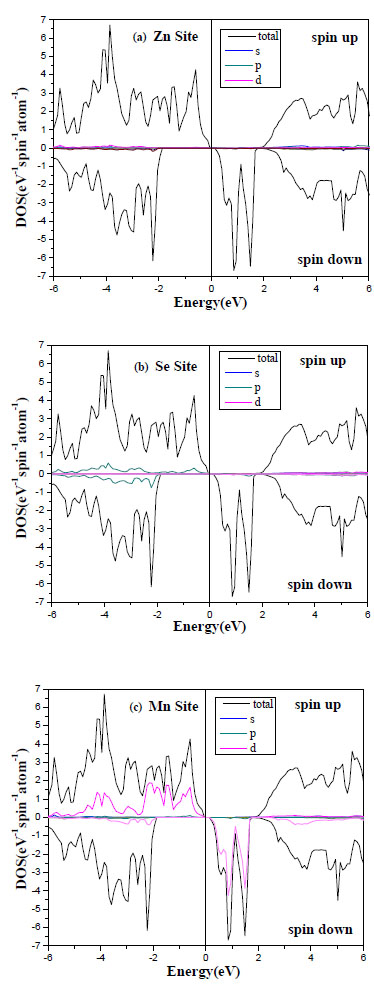 |
Fig. (4) DOS for the ferromagnetic Zn1-xMnxTe for spin up and spin down. |
 |
Fig. (5) DOS for the ferromagnetic Zn1-xMnxTe for spin up and spin down. |
The spin-polarized band structures of ferromagnetic ZnMnTe, ZnMnS and ZnMnSe compounds at the calculated lattice constants are depicted in Figs. (3-5).
The figures show for each compound, the band structures corresponding to spin up and down alignments. We have referenced the zero energy at the top of the valence band for spin up. The bottom of the conduction bands and the top of the valence bands are at the Г point in the Brillouin zone, it is well known that the calculations of the local densities of states (DOS) serve as a qualitative description of the atomic and orbital origins of the various band states. We illustrate our calculations of DOS in Figs. (3-5) for ZnMnS and ZnMnSe, ZnMnTe, respectively. The spin-dependent density of states (DOS) for the ferromagnetic phase is presented in Fig. (2, upper panel). The lower panel presents the contributions to the DOS from the Mn-3d orbitals. We can see that the upper valence band complex has Mn d and Te p characteristics, which differ widely for spin up and spin down. On the other hand, there strong contribution of the Mn spin down states to the valence band. We observe also that the spin up Mn d band is occupied (Figs. 3c, 4c , 5c), and cantered at , 4c) and 5(c)), and centred at
, 5c), and cantered at , 4c) and 5(c)), and centred at
We summarize in Table 2 our results of ΔEc, ΔEv, N0α and N0β for both ZnMnY (Y = S, Se, Te).
Magnetic Properties
Our calculated total and local magnetic moments as well as that of the interstitial site are given in Table 3. It is expected in Mn doped DMSs [29] that: (i) the number of valence minority spin electrons is not changed by the Mn impurity and (ii) each Mn impurity adds five additional majority spin states to the valence band. In Table 3 we show that this description is also valid for ZnMnTe and ZnMnSe and ZnMnS.
Calculated Magnetic Moments (in Bohr Magneton μB of Several Sites and the Total Magnetic Moment for Each Mn for the Ferromagnetic ZnMnY (Y = S, Se, Te) with 25%

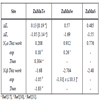
The total magnetic moment of materials ZnMnY(Y = S, Se, Te) equal ≈5μB (μB is the Bohr magneton).
From Table 3 we can see that the p–d hybridization is found to reduce the local magnetic moment of Mn.
We observed the small local magnetic moments on the nonmagnetic S, Se, Te and Zn sites.
CONCLUSION
We have presented in this paper the structural and electronic properties and magnetic moments of the ZnMnY (Y = S, Se, Te), with 25% Mn in the ferromagnetic phase. For FP-LMTO method within the LSDA approximation for exchange-correlation potential.
We have calculated the composition dependence of the lattice constant, bulk modulus, the bulk modulus for alloy increase with the Mn concentration.
We have also determined the exchange constants N0α and N0β . The calculations of total and local magnetic moments have shown that every Mn impurity adds no hole carriers to the perfect ZnY(Y = S, Se, Te) crystal, and that the total magnetic moment is equal to 5μB.