- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Condensed Matter Physics Journal
(Discontinued)
ISSN: 1874-1924 ― Volume 4, 2013
Acentric Nanostructured Assembly as a Strategy for the Design of Organic Electrooptic Materials
Carmine Coluccini , Dario Pasini*
Abstract
The rational utilization of the paradigms of self-assembly and self-organization, essential requisites for the building up of the architectural complexity found in natural systems, is becoming an unavoidable strategy in the world of nanosciences. Recent developments in the field of organic materials for second-order nonlinear optics have focused in the recent past not only towards the optimization of the molecular engineering translating into ultrahigh molecular hyperpolarizabilities, but also, and more fundamentally, towards the realization of acentric assemblies of such chromophores into nanomaterials with high electrooptic responses. Selected, recent examples dealing with these concepts are discussed.
Article Information
Identifiers and Pagination:
Year: 2008Volume: 1
First Page: 7
Last Page: 12
Publisher Id: TOCMPJ-1-7
DOI: 10.2174/1874186X00801010007
Article History:
Received Date: 31/3/2008Revision Received Date: 23/5/2008
Acceptance Date: 27/5/2008
Electronic publication date: 9/6/2008
Collection year: 2008
open-access license: This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.5/), which permits unrestrictive use, distribution, and reproduction in any medium, provided the original work is properly cited.
* Address correspondence to these authors at the Department of Organic Chemistry, University of Pavia, 27100 Pavia, Italy; Tel: +39-0382-987835; Fax: +39-0382-987323; E-mail: dario.pasini@unipv.it
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 31-3-2008 |
Original Manuscript | Acentric Nanostructured Assembly as a Strategy for the Design of Organic Electrooptic Materials | |
INTRODUCTION
The possibility to change the frequency, polarization, and propagation characteristics of optical beams is of great importance for electronic and photonic applications in materials science. Harmonic generation adds tunability to coherent laser sources and allows covering otherwise forbidden spectral ranges. Nonlinear optical (NLO) materials have been proven to be very useful for a variety of fields, spanning from modulation of optical signals, to microfabrication, sensing and medical imaging [1, 2]. In modern telecommunications systems it is fundamental to develop high-speed components for broadband, all-optical signal/information processing. The core component of such technologies is the electro-optic modulator, in which electrical signals are commuted into fiber-optic transmissions. A significant challenge therefore is to obtain materials with large electro-optic responses, which could be readily fabricated into devices at low cost [3].
Large efforts in fundamental research have been put in the last decades in order to continuously improve the performances of second order NLO materials (materials which possess an appreciable quadratic susceptibility). A high-intensity light field produces an induced dipole µi in a molecule, given by the equation:
µi is the ith component of the induced dipole moment, E are the components of the applied electromagnetic field, αij, βijk, γijkt are components of the linear polarizability, the first hyperpolarizability and the second hyperpolarizability tensor respectively [4]. The first hyperpolarizability β is responsible for second order non linear effects as second harmonic generation, frequency mixing, optical rectification and electro-optic effects, and it nullifies in a centrosymmetric environment. Hyperpolarizability β is experimentally determined by EFISHG (Electric Field Induced Second Harmonic Generation) technique or hyper-Rayleigh scattering technique.
In the past, the most common workhorse materials in this field have been inorganic materials, the most relevant being the perovskite lithium niobate. However, organic materials generally possess a better processability than inorganic materials and for this reason polarizable, noncentrosymmetric organic molecules are recently attracting an ever increasing attention. Another important parameter for the evaluation of NLO crystals, together with their NLO properties, is based on their ability to withstand high-power laser sources; in this context, recent studies have shown that organic materials generally possess higher damage thresholds than inorganic crystals [5].
Noncentrosymmetric, dipolar molecules in which polarization in one direction of the molecule is easier than in the opposite direction constitute an important class of molecules with hyperpolarizability properties. This kind of organic molecules are designed after the “push-pull” concept, in which donor and acceptor groups endcap an aromatic, delocalized π-electron system. The degree of ground-state polarization, and therefore the degree of charge separation, is dependent primarily on the chemical structure of the chromophore, but also on the polarity of the medium. Extensive synthetic efforts have been devoted to the optimization of both donor and acceptor groups, and to the introduction of moieties within the π-electron conjugated bridge, which could provide a optimal trade-off between enhancing charge transfer, preventing aggregation, and maintaining good thermal and photochemical stabilities.
If the molecular design aims at the maximization of the intrinsic hyperpolarizability, structural features that translate the molecular NLO response into the bulk phase of the materials are needed. Several strategies have been reported for arranging NLO molecules into materials with high and stable second-order nonlinear optical activities. These are based on chromophore modules incorporation in thin films, in the construction of multilayers, and in functionalized polymers. In polymeric structures, the chromophore alignment needed to optimize the EO response is usually achieved by poling, i.e. by application of an external electric field [6]. Even though these polymeric systems have still limitations (limited thermal stability, tendency to lose chromophore alignment with time), they are still amongst the most practical chemical tools for constructing effective EO solid-state devices [7]. New types of poling schemes, such as post-poling crosslinking strategies in order to lock chromophore alignment, have also been reported [8]. Investigations dealing with the use of modern macromolecular architectures, such as dendrimers, as both hosts for dipolar molecules, or covalent supports for the multivalent organization of active chromophores in the bulk, have recently emerged [9]. The use of specific noncovalent interactions, engineered in such a way that the structure-directed assembly of the chromophores into acentric, oriented materials results in the efficient translation of the molecular electrooptic properties into the bulk phase, has not yet been fully explored, and will be the leading concept reviewed in this paper, with selected recent examples.
DISCUSSION
“State of the Art” Design Principles for Efficient Organic EO Chromophores
As previously stated, the vast majority of efficient organic chromophores are D-π-A (usually dubbed “push-pull”) moieties, i.e. organic compounds with an electron acceptor and an electron donor group endcapping a conjugated π-electron system. An intramolecular charge-transfer (ICT) transition from the polar ground state to the first, charge-separated excited state produces effective polarization along the π-conjugated vector; the classical “two-state model” correlates the molecular hyperpolarizability β, through a proportionality constant k, with the energy gap between the two states (ΔEg), the transition dipole moment between the two states (µg), and the difference in dipole moment between the two states (Δµg) [10]:
Evolving from relatively simple “push-pull” chemical structures such as p-nitroaniline or 4-dimethylamino-4’-nitrostilbene (DANS), which are still used as benchmark compounds to evaluate NLO properties of other molecules, molecular engineering of the chemical backbone has yielded single chromophores with second-order NLO responses, usually quoted as µβ values (µ being the molecular dipole moment), as high as 18000 x 10-48 esu (structures 1 and 2 in Fig. 1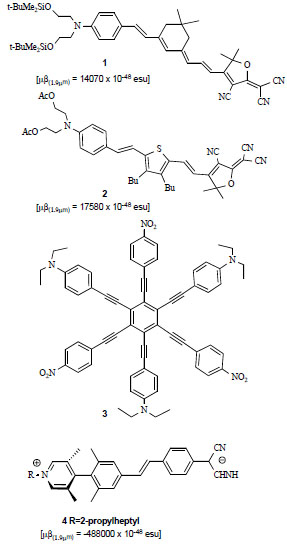 ) [11].
) [11].
 |
Fig. (1) Selected examples of organic chromophores possessing ultrahigh hyperpolarizabilities. |
These compounds afforded an optimal trade-off between the NLO response and transparency issues: the presence, unavoidable in large conjugated systems, of bathochromic shifts in the absorption spectra of the compounds leads to an erosion of the transparency window in the near-IR wavelength region, which is the operational region for many photonic applications; these compounds also show excellent thermal and photochemical stabilities. An improvement in transparency and stability has been recently achieved by using multidimensional ("octopolar") charge-transfer chromophores, such as 3 [12] in Fig. (1) [13]. In octopolar NLO compounds, non-centrosymmetric molecules with no dipole moment, however, a significant increase in second order NLO performance, when compared to chromophores designed after the “traditional” push-pull approach, has not been achieved to date.
A new paradigm for the optimization of molecular hyperpolarizabilities β has been recently proposed by the use of "twisted" D-π-A chromophores [14]. These molecular modules (compound 4 in Fig. 1) consist of a highly polar, charge-separated zwitterionic ground state, and of a less polar, quinonoid-like excited state, thus µβ values exhibited negative signs. Insightful studies, performed by optical, NMR and IR spectroscopies revealed how these unconventional chromophores possess a strong negative solvatochromism, and exhibit a strong tendency for centrosymmetric aggregation in concentrated solutions. Exceptional molecular hyperpolarizabilities (up to µβ = -488000 x 10-48 esu at 1907 nm, measured with the EFISH technique in diluted solutions) have been reported for these systems and, when embedded as hosts in guest polymers subjected to poling, these chromophores produced devices with a very large electro-optic coefficient (r33) of ca. 330 pm/V [15].
Noncovalent Assembly as a Tool for Enhancing EO Response in the Bulk Material
The exploitation of novel strategies aiming at the efficient transfer of the optimized molecular EO properties to the macroscopic level is an active area of research. Although several strategies (polymer poling, Langmuir Blodgett films, etc.) have been briefly commented and referenced above, selected novel approaches, based on the use of weak, noncovalent interactions able to stabilize nanoscopic, acentric ordering in the material will be commented. Dipolar organic compounds with high hyperpolarizability can in principle assemble in noncentrosymmetric (acentric) structures, but electrostatic interactions favor thermodynamically antiparallel arrangement (Fig. 2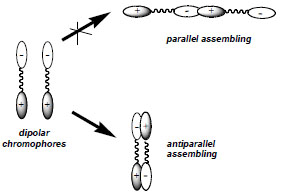 ), thus losing orientation of the chromophores and a strong, macroscopic NLO effect.
), thus losing orientation of the chromophores and a strong, macroscopic NLO effect.
 |
Fig. (2) Thermodynamically favored antiparallel assembling by electrostatic interactions of dipolar compounds. |
Würthner et al. recently proposed a supramolecular approach in which a receptor is employed to orient two merocyanine dyes in the desirable head-to-tail parallel fashion through multiple hydrogen bonding. The design makes use of the organic functionalization of the dyes with complementary recognition groups, as illustrated in Fig. (3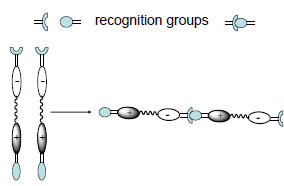 ) [16].
) [16].
 |
Fig. (3) Parallel assembling of dipolar compounds can be obtained by the use of a complementary recognition groups-driven process. |
Merocyanine dyes such as 6 in Fig. (4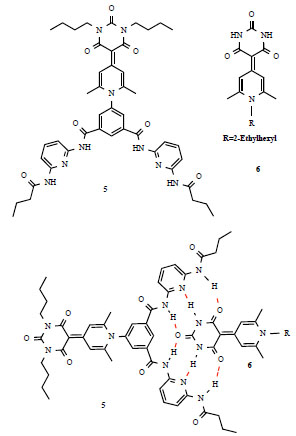 ) are amongst the simplest yet efficient chromophores for NLO applications [4]. Organic functionalization of such substructures afforded compound 5, containing a complementary and suitable binding niche for the supramolecular binding of 6. Head to tail interactions in such compounds is driven by six hydrogen interactions, highlighted in red. The complex 5-6 has been characterized and its thermodynamic strength quantified by NMR spectroscopy. By monitoring the downfield shifts of the NH protons in diacyl pyridine units of 5 during the titration of 5 with 6 and vice versa, it was possible to obtain the binding constant for the formation of 1:1 complex in chloroform (Ka>106M-1) and in dioxane [Ka=(9±3)x103M-1]. Solubility problems in less polar solvents hampered the determination of binding constants in such solvents. The UV/Vis absortion spectra of 5 shows a maximum at λ=396 nm and at λ=379 nm for 6. The spectrum of complex 5-6 indicates additive behavior with two maxima at 380 nm and 389 nm. Another indication of the simple additive behavior, with respect to the absorption properties of the two species in solution, is that the sum of integral absorptions of 5 and 6 (which are proportional to the square of the electric transition dipole moment) matches that of the 1:1 complex 5-6. However, extinction absorption coefficients revealed the presence of mutual chromophore interactions upon complexation, and the variation of the band structure could be associated to the polarization of 6 within the hydrogen bond network, with a weak exciton coupling observed between two chromophores closely positioned in the complex.
) are amongst the simplest yet efficient chromophores for NLO applications [4]. Organic functionalization of such substructures afforded compound 5, containing a complementary and suitable binding niche for the supramolecular binding of 6. Head to tail interactions in such compounds is driven by six hydrogen interactions, highlighted in red. The complex 5-6 has been characterized and its thermodynamic strength quantified by NMR spectroscopy. By monitoring the downfield shifts of the NH protons in diacyl pyridine units of 5 during the titration of 5 with 6 and vice versa, it was possible to obtain the binding constant for the formation of 1:1 complex in chloroform (Ka>106M-1) and in dioxane [Ka=(9±3)x103M-1]. Solubility problems in less polar solvents hampered the determination of binding constants in such solvents. The UV/Vis absortion spectra of 5 shows a maximum at λ=396 nm and at λ=379 nm for 6. The spectrum of complex 5-6 indicates additive behavior with two maxima at 380 nm and 389 nm. Another indication of the simple additive behavior, with respect to the absorption properties of the two species in solution, is that the sum of integral absorptions of 5 and 6 (which are proportional to the square of the electric transition dipole moment) matches that of the 1:1 complex 5-6. However, extinction absorption coefficients revealed the presence of mutual chromophore interactions upon complexation, and the variation of the band structure could be associated to the polarization of 6 within the hydrogen bond network, with a weak exciton coupling observed between two chromophores closely positioned in the complex.
 |
Fig. (4) Parallel orientation in solution of merocyanine NLO dyes. Specific hydrogen bonding interactions are highlighted in red. |
EOA (Electro optic absorbance) spectra, which are absorbance spectra in presence of an electric field and using polarized light, were used to further characterize the electronic structure of the complex. EOA spectra were run for compound 5, 6 and complex 5-6 with light polarized parallel to the electric field and with light polarized perpendicular to electric field. An increased absorbance was observed for the complex when the light beam was polarized parallel to the direction of the electric field, whereas a decrease in absorbance was detected when the direction was orthogonal to the electric field. Furthermore, the effect was far more pronounced in the case of the complex 5-6. The enhancement of absorbance in equimolar mixture confirmed cooperative orientation of dyes in the presence of an electric field. Thus, the judicious use of specific and complementary noncovalent interactions has demonstrated unprecedented degree of dye alignment for this type of chromophore in solution.
The utilization of octopolar NLO chromophores, possessing an inherent push-pull structure but no net dipole moment, is a promising strategy to overcome, at the molecular level, the unavoidable problem of the thermodynamically favorable aggregation tendency of dipolar molecules in concentrated, condensed or bulk phases (see also Fig. 2). Verbiest, Serrano and Heinrich have recently reported the synthesis and characterization of C3- symmetrical octopoles, shown in Fig. (5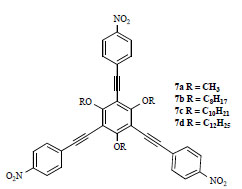 ) [17]. All the compounds showed a broad absorption around 350 nm, testifying for an efficient intramolecular charge-transfer interaction, and a low fluorescence, which facilitates determination of NLO properties using hyper-Rayleigh scattering. Compounds 7b-d, bearing long alkoxy chains, are effective mesogens exhibiting discotic (columnar or nematic) liquid crystalline behavior with quite marked differences in their temperature range intervals.
) [17]. All the compounds showed a broad absorption around 350 nm, testifying for an efficient intramolecular charge-transfer interaction, and a low fluorescence, which facilitates determination of NLO properties using hyper-Rayleigh scattering. Compounds 7b-d, bearing long alkoxy chains, are effective mesogens exhibiting discotic (columnar or nematic) liquid crystalline behavior with quite marked differences in their temperature range intervals.
 |
Fig. (5) Octopolar NLO chromophores functionalized with alkyl chains of varying lengths in order to induce liquid crystalline discotic behavior. |
The hyperpolarizability values were found to be high for all these molecules, and in accordance with previously reported similar octopolar moieties. NLO devices were constructed, in the case of the compounds exhibiting liquid crystalline behavior, such as 7c, by heating a small amount of solid 7c between two glass slides above the melting point, followed by rapid cooling to room temperature. The sample was then placed at the heating stage for measurements of second-harmonic intensity versus temperature, and, as expected, SHG rose drastically (one order of magnitude) in the temperature range associated with liquid crystalline behavior, clearly demonstrating an acentric supramolecular order of the material. This study represents the first example of a simple, efficient NLO device built with octopolar compounds, with responses which are comparable to those of conventional poled-polymer systems.
The groups of Marks and Pagani have recently reported on the realization of heteroaromatic organic chromophores that can be self-organized from the vapor phase into intrinsically acentric, micrometer-scale films [18], (with second-order susceptibilities up to 100 pm/V). The devices are thermally stable and conveniently built in a few hours. Supramolecular acentricity is achieved without electric field poling, and enforced by head-to-tail hydrogen-bonding engineered in the molecules. Films formed by compounds 8-9 (Fig. 6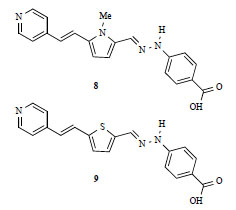 ) were prepared with a combination of covalent chemistry to fix the first layer of chromophores onto glass supports, and of a vapor deposition process to build up the ordered multilayer structure. Compounds 8 and 9 have been engineered in such a way that a head-to-tail orientation can be achieved via hydrogen bonding between the pyridine atom (acceptor) and the carboxylic moiety (donor). The resulting films were transparent at the NLO measurement wavelenght, and of excellent quality, with the electrooptic response of films made using chromophore 8 being two orders of magnitude higher than that of previously reported hydrogen-bonded films.
) were prepared with a combination of covalent chemistry to fix the first layer of chromophores onto glass supports, and of a vapor deposition process to build up the ordered multilayer structure. Compounds 8 and 9 have been engineered in such a way that a head-to-tail orientation can be achieved via hydrogen bonding between the pyridine atom (acceptor) and the carboxylic moiety (donor). The resulting films were transparent at the NLO measurement wavelenght, and of excellent quality, with the electrooptic response of films made using chromophore 8 being two orders of magnitude higher than that of previously reported hydrogen-bonded films.
 |
Fig. (6) Hydrogen-bonded systems form self-organized films by a vapor deposition process. |
As an alternative to hydrogen bonding, halogen bonding is a rather new tool in the supramolecular basket, and can be briefly defined as any noncovalent interaction involving halogens as electrophilic sites. Resnati, Metrangolo and Cariati have recently reported organic chromophores which undergo halogen-bonding mediated assembly in order to tune their NLO response. Structures 10 and 11 (Fig. 7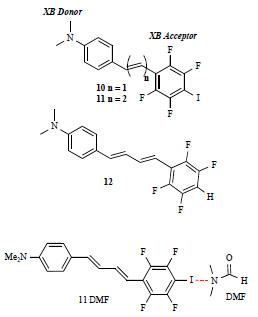 ) are compounds composed of aromatic tetrafluorosubstituted halogen-bond donor (the terminal iodine) and aromatic halogen bond acceptor (the dimethylaniline unit), endcapping a π-electron conjugated system. They self organize in the solid state with head to tail interactions because of halogen bonding between iodine atoms and nitrogen atoms. Compound 12 was synthesized and tested as a model compound, since it hasn’t a terminal iodine atom and cannot give halogen bonding, even though it possesses a DπA structure similar to that of 10 and 11.
) are compounds composed of aromatic tetrafluorosubstituted halogen-bond donor (the terminal iodine) and aromatic halogen bond acceptor (the dimethylaniline unit), endcapping a π-electron conjugated system. They self organize in the solid state with head to tail interactions because of halogen bonding between iodine atoms and nitrogen atoms. Compound 12 was synthesized and tested as a model compound, since it hasn’t a terminal iodine atom and cannot give halogen bonding, even though it possesses a DπA structure similar to that of 10 and 11.
 |
Fig. (7) Halogen-bonded (XB) systems. |
The UV-Vis spectra of 10, 11, 12 in CHCl3 show maxima at 382, 406, 392 nm respectively. Calculations showed that the HOMO and LUMO, both π-orbitals, are extended throughout the molecules, confirming an extended conjugation. The transition is associated with a charge-transfer interaction from a more polar ground state to a less polar excited state [19].
In agreement with the two state model mentioned above, positive values of hyperpolarizability in CHCl3 were observed (µβλ for 10, 11, 12 are +124, +192, +173 x 10-48 esu, respectively). However, measurements of hyperpolarizability in N,N'-Dimethylformamide (DMF) revealed a positive value for 12 (+70 x 10-48 esu), while a strongly negative value for 10 and 11 (-380 and -465 x 10-48 esu, respectively). DMF is an halogen bond acceptor moiety, and can therefore compete with the NMe2 unit. The UV-Vis spectra in DMF of the three compounds were superimposable to those in CHCl3, showing that the nature of HOMO-LUMO transition doesn’t change by changing the solvent. Measurements of hyperpolarizability in CHCl3 in presence of Lewis bases capable of interact with halogen bonding as DMF, piperidine, and quinuclidine (in large excess with respect to compound 11) gave values of -103, -118, -487 x 10-48 esu respectively. The stronger the halogen-bonding acceptor Lewis base, the more negative hyperpolarizability becomes. In agreement with the computational data, the experimental data show how the electrooptic response of these relatively simple push-pull compounds can be tuned, and its sign inverted, by using of complementary molecules (bases) capable of interacting with the substrate via halogen bonds. The hyperpolarizability sign inversion can be explained by the simultaneous presence of two competitive electron-donor sites, acting either intermolecularly or intramolecularly.
Since second harmonic generation (SHG) is only detectable when the bulk medium is non-centrosymmetric; the breaking of inversion symmetry, unavoidable in chiral systems, ensures nonzero second-order nonlinear response. A fully-implemented realization of this concept can be found in the work of Verbiest, Katz et al. [20]. The helicene used (a chiral organic molecule) has been studied both as a single enantiomer and as a racemic mixture (Fig. 8A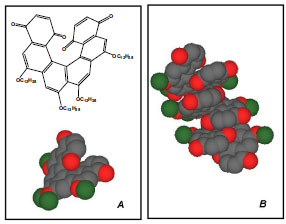 ). Only the enantiomerically-pure form of the compound is able to organize into long fibers, visible under a simple, optical microscope; these fibers are composed as columnar stacks of molecules, as briefly indicated in Fig. (8B), stabilized by noncovalent interactions. This chiral, ordered superstructuring has been confirmed also in solution of apolar solvents, by the use of CD spectroscopy. For the second-harmonic generation studies, films were prepared using the Langmuir-Blodgett technique, revealing excellent optical qualities. From a comparison between the result obtained with racemic and nonracemic films, the nonlinearity seems to arise from the chiral tensor components; furthermore, although the chemical design does not fulfill an optimized "push-pull" structure, the NLO response is high (ca. 50 pm/V). This data suggests that further improvements, with these and other chiral molecular modules, can be achieved through improved molecular engineering and chiral nanostructuring.
). Only the enantiomerically-pure form of the compound is able to organize into long fibers, visible under a simple, optical microscope; these fibers are composed as columnar stacks of molecules, as briefly indicated in Fig. (8B), stabilized by noncovalent interactions. This chiral, ordered superstructuring has been confirmed also in solution of apolar solvents, by the use of CD spectroscopy. For the second-harmonic generation studies, films were prepared using the Langmuir-Blodgett technique, revealing excellent optical qualities. From a comparison between the result obtained with racemic and nonracemic films, the nonlinearity seems to arise from the chiral tensor components; furthermore, although the chemical design does not fulfill an optimized "push-pull" structure, the NLO response is high (ca. 50 pm/V). This data suggests that further improvements, with these and other chiral molecular modules, can be achieved through improved molecular engineering and chiral nanostructuring.

CONCLUSIONS
The design of modern electrooptic devices is certainly an interdisciplinary effort, involving chemists, physicists, material scientists and engineers. It is clear that the paradigms of self-assembly and self-organization can be of great utility in the design of the next generation materials, thus meeting the demands and expectations of an intense research field.
ACKNOWLEDGEMENTS
We gratefully acknowledge funding from the University of Pavia, MIUR (PRIN 2004), Regione Lombardia (INGENIO postdoctoral fellowship to CC) and Fondazione CARIPLO (project "Self-Assembled Nanostructured Materials: A Strategy for the Control of Electrooptic Properties" 2007-2009).