- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Systems Biology Journal
(Discontinued)
ISSN: 1876-3928 ― Volume 5, 2014
Systems Biology Approach for Mapping TNFα-NFκB Mathematical Model to a Protein Interaction Map
Mahesh Visvanathan*, 1, Christian Baumgartner2, Bernhard Tilg 2, Gerald Henry Lushington1
Abstract
We investigated different mathematical models concerning signaling pathways and built a new pathway model for TNFα-NFκB signaling using an integrative analytical approach. This integrative approach consists of a knowledgebase, model designing/visualization and simulation environments. In particular, our new TNFα-NFκB signaling pathway model was developed based on literature studies and the use of ordinary differential equations and a detailed protein-protein interaction connectivity map within this approach. Using the most detailed mathematical model as a base model, three new proteins -- TRAF1, FLIP, and MEKK3 -- were identified and included in our new model. Our results show that this integrative approach offers the most detailed and consistent mathematical description for TNFα-NFκB signaling and further increases the understanding of TNFα-NFκB signaling pathway. This tool can be downloaded through the following link (http://sourceforge.net/projects/dmsp11/).
Article Information
Identifiers and Pagination:
Year: 2010Volume: 3
First Page: 1
Last Page: 8
Publisher Id: TOSYSBJ-3-1
DOI: 10.2174/1876392801003010001
Article History:
Received Date: 3/11/2009Revision Received Date: 1/1/2010
Acceptance Date: 1/3/2010
Electronic publication date: 29/4/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Bioinformatics Core Facility, University of Kansas, Lawrence, KS 66047, USA; Tel: 785-864-3337; Fax: 785-864-8141; E-mail: mvisvanathan@ku.edu
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 3-11-2009 |
Original Manuscript | Systems Biology Approach for Mapping TNFα-NFκB Mathematical Model to a Protein Interaction Map | |
INTRODUCTION
Interactions of molecules are essential for almost all cellular functions. Genes and proteins seldom carry out their functions in isolation. They operate through a number of interactions with other biomolecules. Molecular interactions in biological pathways and networks are highly dynamic and may be controlled by feedback loops and forward regulation mechanisms as well as interlinks with other cellular hierarchies. They make experimental elucidation and computational analysis of pathways extremely challenging. Mathematical modeling is becoming increasingly important as a tool to capture molecular interactions and dynamics from high-throughput experiments. Biological pathways and networks are often represented graphically. Ordinary differential equations (ODEs) have been commonly used to help explain the kinetic process of association and disassociation among molecules in chemical or biochemical reactions.
Tumor necrosis factor-alpha (TNFα) is a cytokine involved in systematic inflammation and is a member of a group of cytokines that all stimulate the acute phase reaction. Dysregulation and over-production of TNFα have been implicated in the pathogenesis of a wide spectrum of human diseases, e.g. sepsis, diabetes, cancer, osteoporosis, multiple sclerosis and crohns disease. Eliminating TNFα by a specific monoclonal antibody, e.g. infliximab, caused dramatic effects on the phenotype of the diseases, with severe side effects. To find specific ways with least side effects to block or partially block TNFα actions, we need to learn more about the signaling pathways of TNFα. Some research efforts have been shifted from intercellular to intracellular signaling in order to increase the knowledge about the involved cell proteins and to get a better understanding of the molecular dynamics of the TNFα-related pathways, especially the TNFα-NFκB signaling pathway.
Biological cartoons and analytical pathway models have been constructed for the TNFα-NFκB signaling pathway respectively. These models can be used for computer simulation studies in order to get insights on setting various experimental scenarios. The better understanding of diseases makes the drug development more appropriate with minimal side effects [1, 2]. Reliable information about proteins and molecular interactions of the signaling pathway is needed to improve the structure of a pathway model. It has known that several protein purification methods are not capable of detecting protein activities in natural concentration. Because of protein over-expression, non-natural protein complex building can occur [3, 4] which could cause faults in modeling. Although a significant amount of biological and biochemical literature is now available in this research field, some of which examine only small parts of the pathway and sometimes with methods relying on over-expression situations. In this paper, we introduce an integrative approach to analyze experimental TNFα-NFκB signaling pathway model. We built an integrative database that includes mathematical modeling data, literature information as well as biological data. In particular, the modeling data consists of kinetic constants, kinetic equations and initial concentrations for building mathematical ODE models. The biological data includes descriptions of proteins, protein-protein interactions and other information concerning signaling pathways. All information can be retrieved and visualized within our integrative computational framework. The paper is organized as follows: Section 2 provides our methodology and delimitates the modeling of the TNFα-NFκB signaling pathway based on ODE models and the literature information. Section 3 describes the results obtained from our extended TNFα-NFκB signaling pathway model. Section 4 gives our discussion and conclusion of this work.
MATERIALS AND Methodology
Integrating Heterogeneous Information for Modeling
The immunosuppression associated with neutralization of TNFα through infliximab results in serious adverse effects e.g. systemic tuberculosis, allergic granulomatosis of the lung, or mild leucopenia in patients with active ankylosing spondylitis [5]. Because of these clinical adverse effects, additional information on the mechanisms of the action of infliximab is needed. In order to increase understanding of the mechanism of TNFα, to find more specific drug targets, to minimize adverse effects and to maintain the duration of the treatment, the focus of attention in research has been shifting from intercellular signals such as TNFα, to intracellular signals, such as the signal transduction pathway from the membrane receptors of TNFα to the transcription factors AP1 and NFκB and to apoptosis.
The exact description of the TNFα pathway is no easy task. Various publications on this topic exist, but they lack focus on the interactions of all the proteins in the pathway. Evidence for the uncertainty about the pathway structure is provided by lots of different biological cartoons dealing with TNFα and NFκB [6-16]. Some of these cartoons use quite different proteins. A recently developed method, the TAP (tandem affinity purification) tag strategy [17, 18], seems to overcome some of the problems and opens a new door in large scale experiments. Further problems have been due to the fact that a lot of experiments were based on protein over-expression situations, which can produce non-physiological complex building. This problem is also solved by the TAP tag strategy [6].
The development of a precise mathematical model is a very difficult task. As the biological knowledge and the experimental data used are insufficient, various assumptions have to be made concerning kinetic parameters and concentrations of proteins involved. The first step to produce a mathematical model is to develop a detailed qualitative model or a cartoon outlining participating proteins after collecting biological and experimental data. This qualitative model/cartoon has to be translated into a quantitative mathematical model [19]. There are two principle ways to construct mathematical models to model the kinetics of biochemical reactions: a deterministic formulation based on nonlinear ordinary differential equations (ODEs) for large numbers of molecules and a stochastic formulation rooted in exponential distribution law. Deterministic models have been commonly used as they can be easily applied with existing off-the-shelf computer software programs, while stochastic models are currently getting more attention and being further developed to capture certain randomness nature of molecular interactions [20, 21]. The alternative to stochastic simulation is using Gillespie’s algorithm, which is an event-driven simulation where the time of next event is exponentially distributed is also used.
A pathway cartoon comprises only qualitative information of the biochemical pathway of interest (e.g. the TNFα-NFκB signaling pathway in our work). The proteins in the cartoon are placed in the region where they are situated in the cell, e.g. the receptor is at the cell membrane, the interacting proteins, which may be located in the cell nucleus, are far away. Information about protein interactions and complex building is hidden in the biological cartoons. Such cartoons do not explain molecular dynamics quantitatively, but they provide a schematic visual overview of the overall dynamics and information processing in cellular systems. Mathematical models of a biochemical pathway can show the chronology of protein complex association and dissociation. This is done with respect to the initial concentrations of the proteins and the kinetic parameters that control the simulation and analysis of theses models [22-26]. Protein-protein interaction connectivity maps also provide important information on which proteins are co-purified with distinct target proteins. To compare and integrate the pathway mathematical model with the protein-protein interaction connectivity maps is a challenge. Based on various cartoons of the TNFα-NFκB signaling pathway, we identified the graphical dynamics of protein complex building. A graphic presentation of a qualitative pathway model of the TNFα-NFκB signaling pathway is shown in Fig. (1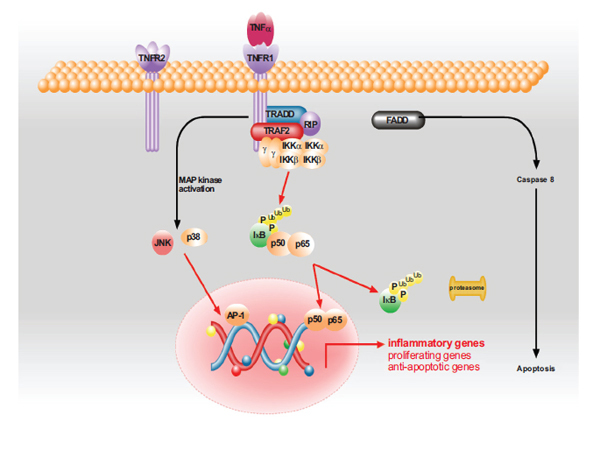 ).
).
 |
Fig. (1) The TNFα pathway model that was developed by our framework, it incorporates biological and modelling knowledge. |
Architecture of the Integrative Framework
Our integrative framework was designed as a 3-tier architecture shown in Fig. (2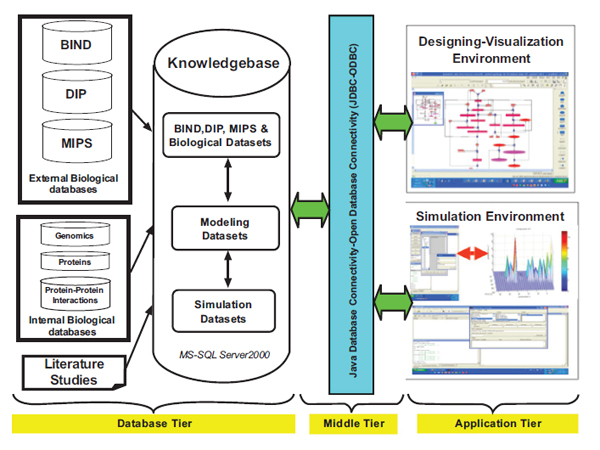 ). It consists of a Java-based pathway designing-visualization environment and a simulation environment in the upper application tier, a Java Database Connectivity-Open Database Connectivity (JDBC-ODBC) in the middle tier, and a relational data management system in the back-end database tier. Within this framework we first designed the TNFα-NFκB pathway model in the designing-visualization environment that allows inclusion of mathematical modeling data, simulation data and biological data from our integrated knowledgebase in the database tier. Then, the designed TNFα-NFκB pathway model was exported in an XML format from the designing environment to the simulation environment.
). It consists of a Java-based pathway designing-visualization environment and a simulation environment in the upper application tier, a Java Database Connectivity-Open Database Connectivity (JDBC-ODBC) in the middle tier, and a relational data management system in the back-end database tier. Within this framework we first designed the TNFα-NFκB pathway model in the designing-visualization environment that allows inclusion of mathematical modeling data, simulation data and biological data from our integrated knowledgebase in the database tier. Then, the designed TNFα-NFκB pathway model was exported in an XML format from the designing environment to the simulation environment.

The organizational structure of the database tier was designed to represent different levels of data. The entities of the knowledgebase contain information grouped into three categories: molecular components, reactions and pathways. These entities inherit both, the biological and modeling information concerning specified pathways. Specifically, the knowledgebase incorporates biological knowledge about components, reactions and pathways from three different online external protein databases: Biomolecular Interaction Network Database (BIND) [27, 28] Database of Interacting Proteins (DIP) [29] and Munich Information Center for Protein Sequences (MIPS) protein-protein interaction database [30] as well as internal experimental verifications and literature studies.
The mathematical modeling knowledge includes kinetic constants, kinetic equations and initial concentrations and is related to the components and reactions of the pathway under investigation (e.g. TNFα-NFκB signaling pathway in our work). In general deterministic formulation, mathematical models of pathways use differential equations relying on fundamental assumptions. For example, a signal transduction system behaves as a slowly time varying non-linear system during a reaction period based on biological observations. Further, it is assumed that a cell keeps the concentration of each signaling protein constant before and after each signaling, that is the concentration of these proteins return to steady state after the reaction. With these assumptions on molecular kinetics, the ordinary differential equations for the mathematical models are derived and stored for simulation of the signaling pathway of interest. We have derived a new ODE model by also incorporating protein-protein interaction connectivity map information for the TNFa-NFκB signaling pathway with our integrative analysis. Simulations and analyses were done using a graphical user interface that was designed and developed using MATLAB®. More details explained in the results section.
RESULTS
For signaling pathway modeling, the following problems occur:
- It is not clear, which proteins should be used to set up a stable model basis, which can easily be extended?
- How is the chronology of protein interactions and which proteins build up a complex?
- How fast and long do the proteins interact (this means, what are the kinetic parameters)?
These problems play a major role in modeling the TNFα-NFκB signaling pathway within our framework. The mathematical models described in [23, 25, 31] related to TNFα pathway showed us that these models contain similar proteins and the general dynamics of complex building was almost identical. Among these models we identified the model based on Cho (we referred it as Model A) as the best available model for possible further improvement in order to address pathway modeling problems mentioned previously. Several other possible extensions/improvements for the pathway Model A were also identified depending on research foci and we concluded that three new proteins – TRAF1, FLIP, and MEKK3 – should be included for possible improvement of Model A. The main focus of our work was to model the TNFα- NFκB signaling pathway starting from TNFα receptor to the transcription factor AP1, rather than going into other levels. By including the three new proteins, we derived an initial extended pathway model using our integrated analytical framework as shown in Fig. (3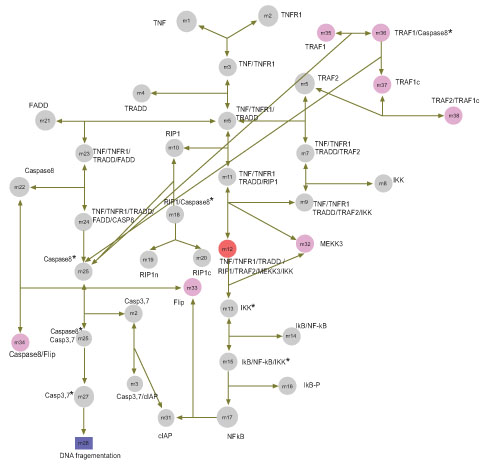 ).
).

It is worth mentioning that TRAF1 and FLIP were also considered in the mathematical models by Schöberl, Ihekwaba and MEKK3 has been intensively discussed [25, 31]. All proteins contained in the cell proliferation module of the extended pathway model were identified and compared among others via the TAP tagged strategy and the protein-protein interaction connectivity map [32]. We noted that some basic interactions, which were modeled, could not be found in the protein-protein interaction connectivity map (i.e. TNFR1-TRADD, TRADD-FADD, TRADD-RIP, RIP-Caspase8 and RIP-IKK). Since cIAP has not been further examined, the interaction with effector Caspases was not modeled. We resulted with a final ODE pathway model (we referred this final ODE pathway model, Model B).
In Model B, several single proteins like TNFR1, TRADD, RIP1, TRAF2, IKK, IκB and NFκB were identified in the protein-protein interaction connectivity map (Fig. 4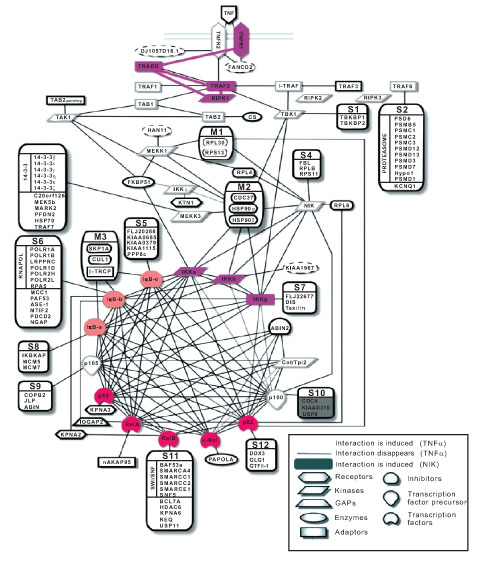 ). The connectivity map comprises no complexes, only individual proteins. IKK is presented by its subunits IKKα, IKKβ and IKKγ, colored lilac, and IκB is presented by its subunits IkBα, IkBβ and IkBε, colored orange. NFκB’s family members (monomers) are colored red and their precursors are mentioned in the connectivity map, i.e. NFκB1 (p50; precursor: p105), NFκB2 (p52; precursor: p100), p65 (RelA), c-Rel (Rel), and RelB. All these seven proteins in both the Model B and the connectivity map. With limited experimental data, further modeling improvements are very difficult. The incorporated biological and modeling knowledge are retrieved from the knowledgebase and integrated into the extended TNFα-NFκB signaling pathway model within our framework.
). The connectivity map comprises no complexes, only individual proteins. IKK is presented by its subunits IKKα, IKKβ and IKKγ, colored lilac, and IκB is presented by its subunits IkBα, IkBβ and IkBε, colored orange. NFκB’s family members (monomers) are colored red and their precursors are mentioned in the connectivity map, i.e. NFκB1 (p50; precursor: p105), NFκB2 (p52; precursor: p100), p65 (RelA), c-Rel (Rel), and RelB. All these seven proteins in both the Model B and the connectivity map. With limited experimental data, further modeling improvements are very difficult. The incorporated biological and modeling knowledge are retrieved from the knowledgebase and integrated into the extended TNFα-NFκB signaling pathway model within our framework.
 |
Fig. (4) The protein-protein interaction connectivity map (Bouwmeester et al. 2004). The proteins that are colored are modeled in our new TNFα-NFκB signaling pathway model B. |
We performed a systematic examination of the TNFα-NFκB signaling pathway by simulating different scenarios to analyze the sensitivity of the Model B with respect to possible changes of the kinetic parameters. The extended TNFa-NFκB signaling pathway model B now including all three new components namely the proteins: MEKK3, FLIP and TRAF1 is indeed quite a stable model. TRAF1 and Caspase8* are reversible from the complex proteins and TRAF1 is irreversibly cleaved and TRAF1c and Caspase8* were yielded. The comparison of the protein concentrations was also performed by plotting the concentrations of the components of other existing mathematical models including model by Cho [24], i.e. Model A, models by Schöberl [31] and Ihekwaba [25] and the new Model B side-by-side. Components, which show extraordinary differences, were examined via further literature investigations. As a result, our extended TNFα-NFκB signaling pathway model B appears to be the most detailed and consistent mathematical model with protein-protein interaction connectivity information incorporated in it.
DISCUSSIONS
Complex interactions of intracellular proteins regarding time course and protein concentrations as well as kinetic behaviors cause high variability in biological functions. To bring light to these complex interaction systems inside of cells, we first have to identify the involved proteins and then to model their interactions by means of sufficient techniques. TNFα is a cytokine, which is implicated in the pathogenesis of various human diseases and therefore it has been under intense investigations for better understanding of the exact signal flow.
Objective of this work was to build a new TNFα- NFκB signaling pathway model that allows integration of protein-protein interaction connectivity map. The mathematical models relating to the TNFα-NFκB signaling pathway contained similar proteins and identical concept behind complex building. Bouwmeester et al. [32] published a connectivity map giving a detailed view on protein-protein interactions of the TNFα-NFκB signaling pathway on a physiological protein concentration level and this map was used, compared and studied with our extended mathematical model.
Mathematical models based on Cho [24], Schöberl [31] and Ihekwaba [25] are available in relation to TNFα- NFκB signaling pathway. Improvements of the existing pathway models are very difficult and challenging. The decision to use the mathematical model by Cho (i.e. Model A) as our base model and extending it was based on the facts that it is the most detailed mathematical model available today for TNFα- NFκB signaling starting from TNFα receptor up to the transcription factor AP1. The Schöberl [24] and Ihekwaba [14] models describe signaling from IKK and NFκB level onwards. The Ihekwaba’s model is concentrating more into the details of modeling IKK interactions rather than the TNFα-NFκB signaling.
Having selected Model A for extension, we found out that this model contained several inconsistencies, regarding the “recycling” of particular proteins after protein complex dissociation for TNFα- NFκB signaling pathway as a whole. Model A did not provide valid reasons why they constructed their model in a particular way. We identified some inconsistencies and corrected the model accordingly with information from the protein-protein interaction connectivity map. In the extended Model B these, inconsistencies were abolished. Hence, this extended model can serve as a more consistent mathematical description to reflect TNFα- NFκB signaling pathway. Our overall integrative analytical approach can also be used to more easily compare and study other future models for further modeling improvements as more experimental data become available. In essence, the knowledgebase of our integrative framework will grow accordingly.
Three new proteins – TRAF1, FLIP, and MEKK3 – were included into the extended model B. TRAF1 and FLIP were already considered in the Schöberl model and MEKK3 has also been intensively discussed in the TNFα- NFκB signaling pathway literature. The importance of this protein has also been pointed out by Bouwmeester et al. [32]. The exact localization of MEKK3 in the signaling pathway and the interactions with other proteins are not fully clear at the moment. That is why the actual position in the extended pathway model has to be regarded as a first approximation to nature.
CONCLUSION
One of the main goals of this work was to use the information of the protein-protein interaction connectivity map to build an improved pathway model. In practice, we found that several protein interactions mentioned in literature could not be found in the connectivity map provided by Bouwmeester et al. [32]. Especially, these were the interactions which are situated in close proximity to the cell membrane. On this regard, the connectivity map just outlines which proteins are parts of the TNFα- NFκB signaling pathway, but does not exactly specify all the occurring interactions between them. To construct a valid mathematical model, it is necessary to have a complete view on the interactions in the signaling pathway. If all the important interactions are revealed, this might require changing the general model structure, as the connectivity map suggests several alternative inhibiting and activating signal flow paths, which have not been modeled yet.
The limitation of experimental data also makes it more challenging to develop a pathway model only for TNFα-NFκB signaling pathway. Information of several sources has to be combined and adapted to fit into one pathway model. Hence, this task makes the possibility of combining models more challenging and interesting. Stochastic mathematical modeling approach is also an interest of our research, and we are currently working on extending a Bayesian probabilistic graphical model framework [21] for TNFα-NFκB signaling pathway. The validation of the structure of TNFα-NFκB signaling pathway by means of experimental data and simulation studies are our on-going work in order to get a more physiologically realistic model. This can be done by incorporating experimental data into the knowledgebase and analyzing the pathway models. It is an iterative interplay between experimental analysis and modeling strategies. Simulation results done on signaling pathways will enable us to interpret the biological system from a global systemic view. This will further increase the understanding of signaling pathways.
ACKNOWLEDGEMENT
This project was supported by Award Number P20 RR016475 from the National Center for Research Resources.