- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




The Open Virology Journal
(Discontinued)
ISSN: 1874-3579 ― Volume 15, 2021
Phylogenesis and Clinical Aspects of Pandemic 2009 Influenza A (H1N1) Virus Infection
Massimo Ciccozzi 1, Muhammed Babakir-Mina 2, 3, Alessandra Lo Presti 1, Fabbio Marcuccilli 2, Carlo Federico Perno 2, 3, Marco Ciotti*, 2
Abstract
During the spring of 2009, a new influenza A (H1N1) virus of swine origin emerged and spread worldwide causing a pandemic influenza. Here, 329 naso-pharyngeal swabs collected from patients with flu-like symptoms were analyzed by real-time PCR for the presence of H1N1 2009 pandemic virus. Twenty-five samples collected from immunocompetent and immunodepressed patients contained the H1N1 pandemic virus. Phylogenetic analysis of the hemagglutinin and neuraminidase genes showed no obvious differences in terms of similarity and/or homology between the sequences identified in immunocompetent individuals and those obtained from immunocompromised patients. Pre-existing clinical conditions may influence the outcome of H1N1 disease.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 5
First Page: 22
Last Page: 26
Publisher Id: TOVJ-5-22
DOI: 10.2174/1874357901105010022
Article History:
Received Date: 29/7/2010Revision Received Date: 6/10/2010
Acceptance Date: 6/10/2010
Electronic publication date: 7/4/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Laboratory of Molecular Virology, Foundation Polyclinic Tor Vergata, Viale Oxford, 81-00133, Rome, Italy; Tel.: +39 06 20902087; Fax: +39 06 20902078; E-mail: marco.ciotti@ptvonline.it
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 29-7-2010 |
Original Manuscript | Phylogenesis and Clinical Aspects of Pandemic 2009 Influenza A (H1N1) Virus Infection | |
INTRODUCTION
In the early spring of 2009, Mexico and the United States reported clusters of human pneumonia cases caused by a novel H1N1 influenza A virus. This virus spread subsequently across the globe at an unprecedented rate, prompting the WHO to declare a pandemic in June 2009. Phylogenetic analysis has inferred that the virus is likely a reassortant between a North American triple-reassortant swine H1N1 or H1N2 virus and a Eurasian lineage H1N1 swine influenza virus [1Dawood FS, Jain S, Finelli L, et al. Novel Swine-Origin Influenza A (H1N1) Virus Investigation Team, Emergence of a novel swine-origin influenza A (H1N1) virus in humans N Engl J Med 2009; 360: 2605-15., 2Garten RJ, Davis CT, Russell CA, et al. Antigenic and genetic characteristics of swine-origin 2009 (H1N1) influenza viruses circulating in humans Science 2009; 325: 197-201.]. The analysis showed that the H1 of this reassortant originated from American pigs, while NA and MP were more likely from European pigs. All of the 2009 isolates appear homogeneous and cluster together, although they are distinct from classical human A (H1N1) viruses [3Babakir-Mina M, Dimonte S, Perno CF, Ciotti M. Origin of the 2009 Mexico influenza virus: a comparative phylogenetic analysis of the principal external antigens and matrix protein Arch Virol 2009; 154: 1349-52.]. Bayesian molecular-clock analysis of each gene of this novel H1N1 virus [4Smith GJD, Vijaykrishna D, Bahl J, et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza an epidemic Nature 2009; 459: 1122-5.] concluded that the mean evolutionary rate is typical of that of swine influenza viruses, but that the duration of unsampled diversity for each gene segment had means that ranged from 9.24 to 17.15 years, suggesting that the proposed ancestors of this virus may have been circulating undetected for nearly a decade. Inadequate surveillance and characterization of influenza A viruses that circulate in swine have been blamed for this evolutionary gap. Also a recent study showed that the domestic birds could act as intermediate hosts of H1N1 reassortants [5Babakir-Mina M, Dimonte S, Ciccozzi M, Perno CF, Ciotti M. The novel swine-origin H1N1 influenza A virus riddle: Is it a domestic bird H1N1-derived virus? New Microbiol 2010; 33: 77-81.]. Multiple genetic groups have been recognized, including one recently predominant lineage [6Valli MB, Meschi S, Selleri M, et al. Evolutionary pattern of pandemic influenza (H1N1) 2009 virus in the late phases of the 2009 pandemic PLoS Curr Influenza 2010; 3: RRN1149.], but any possible clinical importance of different lineages remains uncertain. To date, reassortment has not occurred with human influenza viruses. The level of pulmonary replication of the 2009 H1N1 virus has been higher than that of seasonal influenza A (H1N1) viruses in animals infected experimentally [7Maines TR, Jayaraman A, Belser JA, et al. Transmission and pathogenesis of swine-origin 2009 A (H1N1) influenza viruses in ferrets and mice Science 2009; 325: 484-7.-9Munster VJ, De Wit E, Van den Brand JM, et al. Pathogenesis and transmission of swine-origin 2009 A(H1N1) influenza virus in ferrets Science 2009; 325: 481-3.], but the 2009 pandemicstrain generally lacks mutations that are associated with increased pathogenicity in other influenza viruses [10Padlan EA. The pandemic 2009 (H1N1) swine influenza virus is mild compared to the pandemic 1918 (H1N1) virus because of a proline-to-serine substitution in the receptor-binding site of its hemagglutinin - a hypothesis Med Hypotheses 2010; 74: 240-1.].
Here, a phylogenetic study was carried out on the H1N1 2009 pandemic viruses isolated from immunocompetent and immunocompromised patients admitted at Tor Vergata hospital for flu-like symptoms. Influenza-like illness was defined as the presence of fever >38º C and at least another constitutional symptom (asthenia, headache, malaise, sweating, and chills) and one respiratory symptom (cough, sore throat, nasal congestion).
From September 2009 through March 2010, 329 naso-pharyngeal swabs were collected from patients (127 females and 202 males) admitted at the Polyclinic Tor Vergata for flu-like symptoms. Patient’s age ranged from 1 to 93 years old. The specimens were analysed at the Laboratory of Molecular Virology, Foundation Polyclinic Tor Vergata, Rome, Italy, for the presence of influenza viruses type A and B.
Viral RNA was extracted with the QIAamp RNA mini kit according to the manufacturer’s instruction (QIAGEN S.P.A., Milan, Italy). The RNA was eluted in a volume of 60 µl and stored at 80°- C until analysis. The influenza virus was detected by qualitative real-time PCR using the Influenza virus A/B assay (Cepheid, Sunnyvalle, CA, USA) run on the SmartCycler platform (Cepheid, Sunnyvalle, CA, USA). Influenza A positive cases were re-amplified with primers targeting the hemaglutinin (HA) and neuraminidase (NA) genes of influenza A (H1N1) 2009 pandemic virus. The primer sequences were as follows: F-HA 5’- TGGGGCCATTGCCGGTTTCA-3’ (nucleotide position 1041-1060) and R-HA 5’TGCCCCCAGGGAGACTACCA -3’ (nucleotide position 1647–1628); F-NA 5’-GCCCAGACAAT GGGGCAGTGG-3’ (nucleotide position 590-610) and R-NA 5’-CCGTCTGGCCAAGACCAACCC-3’ (nucleotide position 1379-1359). Reverse-transcription and amplification was carried out using the OneStep RT-PCR Kit according to the manufacturer’s instructions (QIAGEN S S.P.A., Milan, Italy).
The PCR products were analyzed on a 2% agarose gel stained with ethidium bromide and visualized under ultraviolet light. All the necessary precautions were taken to avoid contamination.
The HA and NA amplified fragments were sequenced using the Genome Lab DTCS Quick Start Kit (Beckman Coulter, Fullerton, CA, USA) and run on a Beckman Coulter CEQ 8000 Genetic Analysis System (Fullerton, CA, USA) after column purification. The obtained sequences were submitted to the Genbank and matched against all deposited sequences (http://www.ncbi.nlm.nih.gov/BLAST).
An alignment with a set of reference sequences was obtained using CLUSTAL X [11Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools Nucleic Acids Res 1994; 25: 4876-82.] and manually edited with the Bioedit software [12Hall TA. Bioedit: a user-friendly biological sequence alignment, editor and analysis program for Windows 95/98 NT Nucleic Acids Symp Ser 199; 41: 95-8.]. The evolutionary model was chosen as the best-fitting nucleotide substitution model, according to the Hierarchical Likelihood Ratio Test (HLRT) implemented in the Model Test V3.0 software [13Posada D, Crandall KA. MODEL TEST: Testing the model of DNA substitution Bioinformatics 1998; 14: 817-.]. The statistical robustness and reliability of the branching order within each phylogenetic tree were confirmed by bootstrap analysis using 1000 replicates for the NJ tree and with the Zero Branch Length Test for the Maximum Likelihood (ML) tree. All calculations were performed with PAUP*4.0 software [14Swofford D, Sullivan J. The phylogenetic handbook-A practical approach to DNA and protein phylogeny. New York: Cambridge University Press 2003; pp. 160-206.]. Trees were rooted as mid point root. The accession numbers of the sequences utilized for phylogenetic analysis including those referred to influenza A virus (H1N1) strains identified in our laboratory and labelled A/ROME/PTV1 to 25 are reported in Appendix Table 1.
Twenty-five H1N1 2009 pandemic viruses were identified. Of these, six were from immunodepressed patients (five with hematological malignancies and one HIV-1 infected) and nineteen from immunocompetent individuals (Table 1). A Maximum Likelihood phylogenetic tree was generated for the HA and NA genes (Figs. 1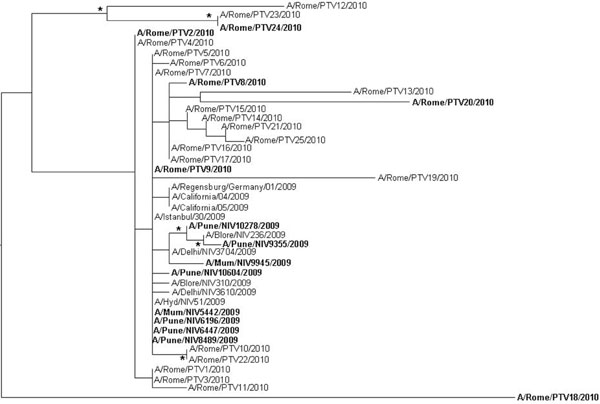 , 2
, 2 ). In the HA tree, a main clade was generated where the strains collected from Italian immunodepressed patients and Indian fatal cases [15Potdar VA, Chadha MS, Jadhav SM, et al. Genetic characterization of the Influenza A pandemic (H1N1) 2009 virus isolates from India PLoS ONE 2010; 5: e9693.] (in bold) intermixed with those obtained from immunocompetent Italian patients or Indian recovered cases (regular font). This is clearly shown by the strains A/Pune/NIV10278/2009 and A/Pune/NIV9355/2009 obtained from two fatal cases which cluster with the strain A/Blore/NIV236/2009 isolated from a recovered patient; and outside the main clade, by the strain A/Rome/PTV24/2010 isolated from an immunocompromised patient which clusters with the strain A/Rome/PTV12/2010 and A/Rome/PTV23/2010 obtained from immunocompetent individuals. Both clusters are statistically supported. A similar picture was observed with the NA tree (Fig. 2). Even in this case the viral strains from immunocompromised or death patients intermixed with those isolated from immucompetent patients with the exception of two small clusters (A/Pune/NIV10604/2009, A/Pune/NIV6196/ 2009 and A/Rome/PTV8/2010, A/Rome/PTV9/2010) which were collected from two fatal cases and immunocompromised patients, respectively.
). In the HA tree, a main clade was generated where the strains collected from Italian immunodepressed patients and Indian fatal cases [15Potdar VA, Chadha MS, Jadhav SM, et al. Genetic characterization of the Influenza A pandemic (H1N1) 2009 virus isolates from India PLoS ONE 2010; 5: e9693.] (in bold) intermixed with those obtained from immunocompetent Italian patients or Indian recovered cases (regular font). This is clearly shown by the strains A/Pune/NIV10278/2009 and A/Pune/NIV9355/2009 obtained from two fatal cases which cluster with the strain A/Blore/NIV236/2009 isolated from a recovered patient; and outside the main clade, by the strain A/Rome/PTV24/2010 isolated from an immunocompromised patient which clusters with the strain A/Rome/PTV12/2010 and A/Rome/PTV23/2010 obtained from immunocompetent individuals. Both clusters are statistically supported. A similar picture was observed with the NA tree (Fig. 2). Even in this case the viral strains from immunocompromised or death patients intermixed with those isolated from immucompetent patients with the exception of two small clusters (A/Pune/NIV10604/2009, A/Pune/NIV6196/ 2009 and A/Rome/PTV8/2010, A/Rome/PTV9/2010) which were collected from two fatal cases and immunocompromised patients, respectively.


The results of this study indicate no obvious differences in viral sequences between the H1N1 pandemic viruses isolated from immunocompetent individuals and those identified in recovered immunocompromised patients and/or fatal cases. We can exclude that the lack of sequence variations observed in Italian immunocompetent and immunocompromised patients was due to contamination of virus sequences since the samples and sequencing were handled appropriately using separate rooms and including controls in each run as also shown by the phylogenetic trees.
Indeed, mutations that have been linked to fatal cases have also been detected in recovered cases [15Potdar VA, Chadha MS, Jadhav SM, et al. Genetic characterization of the Influenza A pandemic (H1N1) 2009 virus isolates from India PLoS ONE 2010; 5: e9693., 16Puzelli S, Facchini M, Spagnolo D, et al. Transmission of hemagglutinin D222G mutant strain of pandemic (H1N1) 2009 virus Emerg Infect Dis 2010; 5: 863-5.]; therefore their role in viral pathogenesis remains unclear. Preexisting specific severe clinical conditions may play an important role in determining the outcome of H1N1disease [17Writing Committee of the WHO Consultation on Clinical Aspects of Pandemic (H1N1) 2009 Influenza. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection N Engl J Med 2010; 362: 1708-9.].