- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




The Open Virology Journal
(Discontinued)
ISSN: 1874-3579 ― Volume 15, 2021
The Invisible Enemy – How Human Papillomaviruses Avoid Recognition and Clearance by the Host Immune System
Agnieszka K Grabowska, Angelika B Riemer*
Abstract
Human papillomavirus (HPV) needs to persist in squamous epithelia for a certain amount of time to complete its reproductive cycle. Therefore, the virus has evolved multiple immune evasion strategies. The interplay of these immune evasion mechanisms with the host immune system decides whether a HPV infection is cleared or becomes persistent. Clearance of HPV-induced lesions is mediated by a cellular immune response, consisting of both cytotoxic T lymphocyte and T helper cell responses. Persistent HPV infection, on the other hand, is the single most important risk factor for the development of HPV-associated premalignant lesions and HPV-driven cancers. This article reviews the immune evasion mechanisms employed by high-risk HPVs to escape host immune recognition and attack.
Article Information
Identifiers and Pagination:
Year: 2012Volume: 6
Issue: Suppl 2
First Page: 249
Last Page: 256
Publisher Id: TOVJ-6-249
DOI: 10.2174/1874357901206010249
Article History:
Received Date: 21/5/2012Revision Received Date: 6/6/2012
Acceptance Date: 15/6/2012
Electronic publication date: 28/12/2012
Collection year: 2012
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at Immunotherapy and -prevention, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany; Tel: +49 6221 42 3820; Fax: +49 6221 42 4852; E-mail: a.riemer@dkfz.de
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 21-5-2012 |
Original Manuscript | The Invisible Enemy – How Human Papillomaviruses Avoid Recognition and Clearance by the Host Immune System | |
INTRODUCTION
Cervical carcinoma and several other malignancies arise as a consequence of persistent infection with high-risk types of human papillomavirus (HPV). Papillomaviruses are small nonenveloped double-stranded DNA viruses with strict tissue and species specificity. Over 190 distinct genotypes have been identified to date, 151 of them isolated from humans [1 Bernard HU, Burk RD, Chen Z, van Doorslaer K, Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments Virology 2010; 401(1): 70-9.]. Papillomaviruses exclusively infect squamous epithelia, i.e. skin and mucosae. Skin types induce common warts, and may be implicated in non-melanoma skin cancer [2 Bouwes BJN, Neale RE, Abeni D, et al. Multicenter study of the association between betapapillomavirus infection and cutaneous squamous cell carcinoma Cancer Res 2010; 70(23): 9777-86.]. The mucosal types of HPV fall in two groups – so-called low-risk types (mainly HPV 6 and 11), which induce genital warts, and the high-risk types, which lead to cervical carcinoma and several other malignancies, such as anal cancer and oropharyngeal carcinomas [3 zur Hausen H. Papillomaviruses in human cancer Appl Pathol 1987; 5(1): 19-24., 4 zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application Nat Rev Cancer 2002; 2(5): 342-50.]. The most prevalent high-risk HPV types are HPV16 and HPV18, being responsible for 50% and 20%, respectively, of cervical cancer cases globally [5 Li N, Franceschi S, Howell-Jones R, Snijders PJ, Clifford GM. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: variation by geographical region, histological type and year of publication Int J Cancer 2011; 128(4): 927-35.]. The extracervical HPV-mediated cancers are almost exclusively caused by HPV16. Taken together, HPV causes 530,000 new cancer cases and 275,000 deaths each year [6 Arbyn M, Castellsague X, de Sanjose S, et al. Worldwide burden of cervical cancer in 2008 Ann Oncol 2011; 22(12): 2675-86.].
Natural history studies indicate that nearly every sexually active individual will acquire at least one high-risk HPV infection during their lifetime [7 Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer Lancet 2007; 370(9590): 890-907., 8 Baseman JG, Koutsky LA. The epidemiology of human papillomavirus infections J Clin Virol 2005; 32(Suppl 1): S16-24.]. Fortunately, the majority of HPV infections are eradicated by the host immune system within 1-2 years of acquisition (median 6 months) [9 Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues Nat Rev Cancer 2007; 7(1): 11-22.], and only <1% of infected people develop HPV-mediated cancers [10 Koutsky L. Epidemiology of genital human papillomavirus infection Am J Med 1997; 102(5A): 3-8., 11 Evander M, Edlund K, Gustafsson A, et al. Human papillomavirus infection is transient in young women: a population-based cohort study J Infect Dis 1995; 171(4): 1026-30.]. Nevertheless, immune clearance of HPV is much slower than clearance of most other viruses, and the fact that some HPV-associated lesions can persist and progress into cancer emphasizes the capacity of HPV to escape host immune surveillance. Detailed understanding of these events on a cellular and molecular level may result in therapeutic strategies to overcome immune escape. This review will focus on the immune evasion strategies employed by HPV. In this context, it is necessary to give an overview of the HPV life cycle first.
The HPV Infectious Life Cycle
HPV exclusively infects the basal layer of epithelial cells, after the basement membrane of a squamous epithelium has been exposed by a microwound [12 Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer Clin Sci (Lond) 2006; 110(5): 525-41.]. The HPV genome consists of approximately 8 kb circular, double-stranded DNA, which is organized into coding and non-coding regions. 6 early open reading frames (ORFs) encode early proteins: E1, E2, E4, E5, E6 and E7; and 2 late ORFs encode late proteins: L1 and L2. The non-coding regulatory region is critical for initiation of viral DNA replication and transcription of the viral genes [12 Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer Clin Sci (Lond) 2006; 110(5): 525-41.-14 Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity Rev Med Virol 2006; 16(2): 83-97.]. The initial phase of HPV infection is characterized by presence of the virus as an episome in the basal layer of undifferentiated epithelial cells, which is maintained only at very low levels (50 to 100 copies per cell) [15 Bedell MA, Hudson JB, Golub TR, et al. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation J Virol 1991; 65(5): 2254-60.]. Viral proteins are expressed at low levels as well and confined mainly to the nucleus of the basal cells [16 Doorbar J. The papillomavirus life cycle J Clin Virol 2005; 32 (Suppl 1): S7-15.]. Viral genome replication requires two viral initiation factors: E1, which contains helicase-ATPase activity, and E2 with multiple activities in replication, transactivation and repression. Because HPV does not encode DNA polymerase activity for viral genome replication, the host DNA replication machinery is required. However, in squamous epithelia, only the basal cells divide and DNA replication activity is normally suppressed in differentiating cells that exit from the basal layer. To hold the cellular replication machinery active, the viral proteins E6 and E7 are produced, which inactivate p53 and the retinoblastoma protein (pRb), respectively, to prevent cell growth arrest and delay differentiation. E7 binds the unphosphorylated form of pRb and forces infected keratinocytes to remain in a proliferative state. Moreover, it stimulates cyclin A and E, promoting G0/G1 progression. The E6 protein induces the rapid degradation of p53 through ubiquitin-dependent proteolysis to prevent apoptosis and allows cell survival [13 Kajitani N, Satsuka A, Kawate A, Sakai H. Productive lifecycle of human papillomaviruses that depends upon squamous epithelial differentiation Front Microbiol 2012; 3: 152., 17 Munger K, Scheffner M, Huibregtse JM, Howley PM. Interactions of HPV E6 and E7 oncoproteins with tumour suppressor gene products Cancer Surv 1992; 12: 197-217.-19 Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation Nat Rev Cancer 2010; 10(8): 550-60.].
The expression of these early viral genes is tightly controlled under the differentiation program of keratinocytes and increased only when keratinocytes migrate through the upper layers of the epithelium. When the infected keratinocytes enter the differentiating compartment in the suprabasal layers of the epithelium, the expression of all viral genes is induced to accelerate viral genome replication, with amplification of viral copy number to thousands of copies per cell [12 Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer Clin Sci (Lond) 2006; 110(5): 525-41., 14 Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity Rev Med Virol 2006; 16(2): 83-97.]. The final stage of HPV infection leads to packing of amplified genomes into infectious particles formed by the L1 and L2 proteins, which spontaneously self-assemble into icosahedral capsids. New virus particles are produced within the uppermost layers of the epithelium, and released by the normal process of epithelial cell shedding at the end of their lifespan [12 Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer Clin Sci (Lond) 2006; 110(5): 525-41.].
IMMUNE EVASION STRATEGIES EMPLOYED BY HPV
Protective immunity results from the interplay of non-specific innate immunity and antigen-specific adaptive immunity. The innate immune system senses “danger” via signals from molecules that would normally not be found in the human body, such as damaged tissue, repetitive surface structures of bacterial cell walls, or DNA sequences containing typical viral sequence motives. These structures are recognized by pattern recognition receptors, such as e.g. Toll-like receptors (TLRs) [20 Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors Nat Immunol 2010; 11(5): 373-84., 21 Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity Cell 2006; 124(4): 783-801.]. Sentinel cells, such as dendritic cells (DCs) or Langerhans cells (LCs) in the skin and mucosa, continuously screen the environment and - if triggered - coordinate innate immune effectors and the initiation of an adaptive immune response [22 Stern PL. Immune control of human papillomavirus (HPV) associated anogenital disease and potential for vaccination J Clin Virol 2005; 32 (Suppl 1): S72-81.]. HPV has evolved mechanisms both to avoid initial recognition and to interfere with adaptive immunity.
Establishment of Immunological Ignorance
The key viral strategy to avoid the host immune system is to maintain a low profile. The virus exclusively infects epithelial cells, and the whole replicative cycle happens outside the basement membrane, away from the dermal immune effector cells.
The virus encodes only non-secreted proteins, which are mostly localized in the nucleus of infected cells and expressed at a very low level. To keep expression of the viral genes low, HPV utilizes two mechanisms: the viral protein E2 acts as a repressor of other early gene expression in basal cells [12 Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer Clin Sci (Lond) 2006; 110(5): 525-41.]. Moreover, HPV exploits the redundancy in the genetic code. The codon usage pattern in the HPV genome differs from the commonly used mammalian codons. The potency of this mechanism has been demonstrated by studies replacing viral codons with codons preferentially used in keratinocytes, which led to significant protein up-regulation [23 Zhao KN, Chen J. Codon usage roles in human papillomavirus Rev Med Virol 2011; 21(6): 397-411., 24 Zhou J, Liu WJ, Peng SW, Sun XY, Frazer I. Papillomavirus capsid protein expression level depends on the match between codon usage and tRNA availability J Virol 1999; 73(6): 4972-82.]. This strategy helps HPV to be invisible to the host immune system at the early stage of viral infection.
Increased viral protein expression only occurs in keratinocytes in the upper layers of the epithelium where the immune system has limited access. Furthermore, there is no viremia, no cell death, and no cell lysis upon viral shedding [16 Doorbar J. The papillomavirus life cycle J Clin Virol 2005; 32 (Suppl 1): S7-15.]. Additionally, HPV E6 and E7 have been implicated in direct inhibition of TLR9-mediated pathways by down-regulating the transcription of the TLR9 gene [25 Hasan UA, Bates E, Takeshita F, et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16 J Immunol 2007; 178(5): 3186-97.].
Thus, HPV infection does not elicit any danger signals, which would be crucial for activation and trafficking of antigen presenting cells (APCs), production of proinflammatory cytokines and initiation of adaptive immune responses [12 Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer Clin Sci (Lond) 2006; 110(5): 525-41., 14 Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity Rev Med Virol 2006; 16(2): 83-97.].
Modulation of Apoptosis
Apoptosis – programmed cell death – is regulated physiologically and genetically, and plays a central role in development, morphogenesis, normal cell turnover and immune system function [26 Hengartner MO. The biochemistry of apoptosis Nature 2000;
407(6805): 770-6., 27 Vermeulen K, Van Bockstaele DR, Berneman ZN. Apoptosis:
mechanisms and relevance in cancer Ann Hematol 2005; 84(10): 627-39.]. Two major apoptotic pathways have been identified, both activating effector caspases. The extrinsic death receptor pathway starts with ligation of a death ligand (CD95L) to its transmembrane death receptor (CD95), followed by activation of caspases in the death-inducing signaling complex. The intrinsic pathway involves mitochondria, which release caspase-activating proteins into the cytosol, thereby forming the apoptosome where caspases will bind and become activated [26 Hengartner MO. The biochemistry of apoptosis Nature 2000;
407(6805): 770-6., 27 Vermeulen K, Van Bockstaele DR, Berneman ZN. Apoptosis:
mechanisms and relevance in cancer Ann Hematol 2005; 84(10): 627-39.]. Apoptosis resistance is an important aspect in viral infection and progression to cancer, because it might contribute to immune escape. Virus-infected or tumor cells can acquire resistance to apoptosis by the expression of anti-apoptotic proteins or by the down-regulation or mutation of pro-apoptotic proteins. Indeed, HPVs have evolved mechanisms to modulate apoptosis to avoid immune attack and establish successful infection [28 Garnett TO, Duerksen-Hughes PJ. Modulation of apoptosis by human
papillomavirus (HPV) oncoproteins Arch Virol 2006; 151(12): 2321-35.] (Fig. 1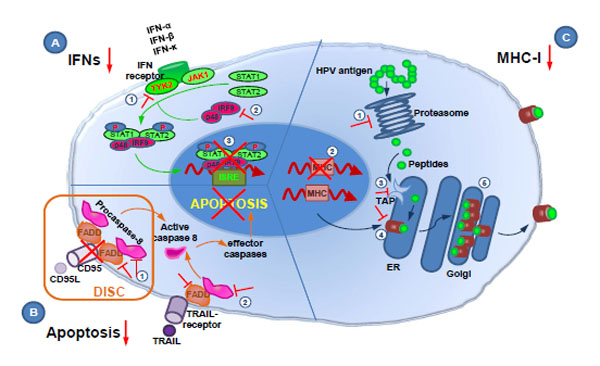 ). The E5 protein inhibits tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)- and CD95L-mediated apoptosis at early stages of HPV infection when the viral genome is episomal [29 Kabsch K, Mossadegh N, Kohl A, et al. The HPV-16 E5 protein
inhibits TRAIL- and FasL-mediated apoptosis in human keratinocyte
raft cultures Intervirology 2004; 47(1): 48-56., 30 Venuti A, Paolini F, Nasir L, et al. Papillomavirus E5: the smallest
oncoprotein with many functions Mol Cancer 2011; 10: 140.]. E5 promotes down-regulation of the CD95 receptor and CD95 surface location, and additionally modulates the formation of the death-inducing signaling complex (DISC) [29 Kabsch K, Mossadegh N, Kohl A, et al. The HPV-16 E5 protein
inhibits TRAIL- and FasL-mediated apoptosis in human keratinocyte
raft cultures Intervirology 2004; 47(1): 48-56., 31 Lagunas-Martinez A, Madrid-Marina V, Gariglio P. Modulation of
apoptosis by early human papillomavirus proteins in cervical cancer
Biochim Biophys Acta 2010; 1805(1): 6-16.]. The E6 protein impairs apoptosis through the ATP-dependent degradation of pro-apoptotic proteins such as p53, FADD, procaspase-8, or c-Myc, employing the ubiquitin-proteasome pathway [32 Gross-Mesilaty S, Reinstein E, Bercovich B, et al. Basal and human
papillomavirus E6 oncoprotein-induced degradation of Myc proteins by
the ubiquitin pathway Proc Natl Acad Sci USA 1998; 95(14): 8058-63.-34 Garnett TO, Filippova M, Duerksen-Hughes PJ. Accelerated
degradation of FADD and procaspase 8 in cells expressing human
papilloma virus 16 E6 impairs TRAIL-mediated apoptosis Cell Death
Differ 2006; 13(11): 1915-26.].
). The E5 protein inhibits tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)- and CD95L-mediated apoptosis at early stages of HPV infection when the viral genome is episomal [29 Kabsch K, Mossadegh N, Kohl A, et al. The HPV-16 E5 protein
inhibits TRAIL- and FasL-mediated apoptosis in human keratinocyte
raft cultures Intervirology 2004; 47(1): 48-56., 30 Venuti A, Paolini F, Nasir L, et al. Papillomavirus E5: the smallest
oncoprotein with many functions Mol Cancer 2011; 10: 140.]. E5 promotes down-regulation of the CD95 receptor and CD95 surface location, and additionally modulates the formation of the death-inducing signaling complex (DISC) [29 Kabsch K, Mossadegh N, Kohl A, et al. The HPV-16 E5 protein
inhibits TRAIL- and FasL-mediated apoptosis in human keratinocyte
raft cultures Intervirology 2004; 47(1): 48-56., 31 Lagunas-Martinez A, Madrid-Marina V, Gariglio P. Modulation of
apoptosis by early human papillomavirus proteins in cervical cancer
Biochim Biophys Acta 2010; 1805(1): 6-16.]. The E6 protein impairs apoptosis through the ATP-dependent degradation of pro-apoptotic proteins such as p53, FADD, procaspase-8, or c-Myc, employing the ubiquitin-proteasome pathway [32 Gross-Mesilaty S, Reinstein E, Bercovich B, et al. Basal and human
papillomavirus E6 oncoprotein-induced degradation of Myc proteins by
the ubiquitin pathway Proc Natl Acad Sci USA 1998; 95(14): 8058-63.-34 Garnett TO, Filippova M, Duerksen-Hughes PJ. Accelerated
degradation of FADD and procaspase 8 in cells expressing human
papilloma virus 16 E6 impairs TRAIL-mediated apoptosis Cell Death
Differ 2006; 13(11): 1915-26.].

Interestingly, recent studies suggest that high-risk HPV proteins E6 and E7 are able to independently activate low levels of caspases upon epithelial differentiation to induce productive viral replication, representing a way by which HPV controls viral gene expression in differentiating cells [35 Moody CA, Fradet-Turcotte A, Archambault J, Laimins LA. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification Proc Natl Acad Sci USA 2007; 104(49): 19541-6.]. HPV-mediated caspase activation is associated with increased levels of anti-apoptotic factors, which may be important in providing a balance between cell viability and cell death upon differentiation. E7-mediated activation of caspases is inhibited in E6-expressing cells and may occur only in the absence of E6 [35 Moody CA, Fradet-Turcotte A, Archambault J, Laimins LA. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification Proc Natl Acad Sci USA 2007; 104(49): 19541-6.]. It is intriguing that HPV can inhibit apoptosis in undifferentiated cells, but influence apoptotic signaling pathways in differentiating cells. It is possible that during a productive viral life cycle both inhibition and induction of apoptosis are required, and that E6 controls apoptosis pathways in both directions, depending on the differentiation stage [31 Lagunas-Martinez A, Madrid-Marina V, Gariglio P. Modulation of apoptosis by early human papillomavirus proteins in cervical cancer Biochim Biophys Acta 2010; 1805(1): 6-16., 35 Moody CA, Fradet-Turcotte A, Archambault J, Laimins LA. Human papillomaviruses activate caspases upon epithelial differentiation to induce viral genome amplification Proc Natl Acad Sci USA 2007; 104(49): 19541-6.]. Nevertheless, this phenomenon still needs to be investigated in more detail.
Dysregulation of Interferon Responses
Interferons (IFNs) are widely expressed cytokines that have potent anti-viral, immunostimulatory, and growth-inhibitory effects. The two main classes of interferons comprise type I IFNs (IFN-α, IFN-β, IFN-κ) and type II IFN (IFN-γ). Type I IFNs are the first line of immune defense against viral infections, causing the so-called “antiviral state” in infected cells, and enhancing immune responses through the stimulation of dendritic cells. This indicates that IFN-α and IFN-β act as an important link between innate and adaptive immunity [36 Le Bon A, Tough DF. Links between innate and adaptive immunity via
type I interferon Curr Opin Immunol 2002; 14(4): 432-6.]. Upon viral infection, a signal transduction pathway involving the TYK2 and JAK1 kinases is activated, leading to a transcription factor complex composed of STAT1, STAT2, and interferon regulatory factor 9 (IRF-9), and the increased production of IFN-α and IFN-β via IRF-3 and IRF-7 [37 Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated
signalling Nat Rev Immunol 2005; 5(5): 375-86.]. Like most DNA viruses, HPV is able to directly modulate the interferon signaling cascade and IFN synthesis (Fig. 1). During infection with high-risk HPV, the E6 protein can inhibit IRF3 transactivation, preventing IFN-β induction [38 Ronco LV, Karpova AY, Vidal M, Howley PM. Human
papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity Genes Dev 1998; 12(13): 2061-72.]. HPV E6 has also been postulated to interfere with the TYK2 kinase, which is required for the activation of IFN-stimulated gene (ISG) transcription. Dysfunction of TYK2 prevents its binding to the IFN receptor and blocks phosphorylation of TYK2, STAT1 and STAT2, thus dysregulating the JAK-STAT activation machinery [39 Li S, Labrecque S, Gauzzi MC, et al. The human papilloma virus
(HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs
Jak-STAT activation by interferon-alpha Oncogene 1999; 18(42): 5727-37.]. The E7 protein is able to inhibit the IRF1 and IRF9 transcription factors, which contribute to ISG transcription. E7 binds to the p48/IRF9 complex and abrogates its translocation to the nucleus, thereby inhibiting the formation of the IFN-stimulated gene factor 3 (ISGF3) transcription complex that binds the IFN-specific response element (ISRE) in the nucleus [40 Barnard P, Payne E, McMillan NA. The human papillomavirus E7
protein is able to inhibit the antiviral and anti-growth functions of
interferon-alpha Virology 2000; 277(2): 411-9.]. Interaction of the E7 protein with IRF1 is characterized by recruiting histone deacetylases to the IFN-β promoter site, thus blocking transcription [41 Perea SE, Massimi P, Banks L. Human papillomavirus type 16 E7
impairs the activation of the interferon regulatory factor-1 Int J Mol
Med 2000; 5(6): 661-., 42 Park JS, Kim EJ, Kwon HJ, Hwang ES, Namkoong SE, Um SJ. Inactivation of interferon regulatory factor-1 tumor suppressor protein
by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis J Biol Chem 2000;
275(10): 6764-9.]. In vivo HPV E7 inhibits the trans-activation function of IRF1, which leads to down-regulation of TAP1 (transporter associated with antigen processing 1), IFN-β, and MCP1 (monocyte chemo-attractant protein-1) [43 Um SJ, Rhyu JW, Kim EJ, Jeon KC, Hwang ES, Park JS. Abrogation
of IRF-1 response by high-risk HPV E7 protein in vivo Cancer Lett
2002; 179(2): 205-12.].
More recently, it was found that high-risk HPV E6 plays a potential role in repression of the constitutive transcription of IFN-κ via reduction of ISG expression [44 Rincon-Orozco B, Halec G, Rosenberger S, et al. Epigenetic silencing of interferon-kappa in human papillomavirus type 16-positive cells Cancer Res 2009; 69(22): 8718-25., 45 Reiser J, Hurst J, Voges M, et al. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression J Virol 2011; 85(21): 11372-80.]. IFN-κ not only regulates antiviral-ISG expression and components of the IFN pathway, but also maintains the expression of pathogen recognition receptors such as TLR3, which in turn control inducible IFN expression in normal human keratinocytes [45 Reiser J, Hurst J, Voges M, et al. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression J Virol 2011; 85(21): 11372-80.]. Taken together, the HPV E6 and E7 proteins directly influence expression and functions of IFNs to prevent host immune attack.
A recent genome-wide expression profiling study in HPV-infected basal keratinocytes [46 Karim R, Meyers C, Backendorf C, et al. Human papillomavirus deregulates the response of a cellular network comprising of chemotactic and proinflammatory genes PLoS One 2011; 6(3): e17848.] revealed that HPV not only influences the interferon pathway, but also dampens a whole network of genes downstream of the pattern recognition receptors (PRRs, such as TLRs and RIG-I-like receptors), including antimicrobial molecules, chemotactic and pro-inflammatory cytokines, the inflammasome, and components of the antigen processing machinery. IL-1β and IL-6 were found to be at the center of this network. The fact that HPV was only found to dampen, but not to completely block, PRR signaling corresponds to the clinical finding that HPV clearance is slow but eventually successful in the majority of infected individuals.
Perturbation of Antigen Processing and Presentation
T cells - both CD8+ cytotoxic T lymphocytes (CTLs) and CD4+ helper T cells (Th) - play a critical role in HPV control and clearance [47 Stanley MA. Immunobiology of papillomavirus infections J Reprod Immunol 2001; 52(1-2): 45-59.]. The major histocompatibility complex (MHC) - in humans known as human leukocyte antigen (HLA) - is a key player in the antigen-specific cell-mediated immune response. These cell surface molecules act as restricting elements in the recognition of antigens by T cells. MHC class I (MHC-I) presents antigen to CTLs. MHC class II (MHC-II) surface expression is restricted to professional antigen presenting cells (APCs), such as macrophages, B-cells and especially dendritic cells (DCs) that present antigen to T helper cells (Th) [48 Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation Cell 1994; 76(2): 287-99.]. Many viruses down-regulate MHC molecules to evade recognition and elimination by the immune system and HPV is not an exception [49 Ashrafi GH, Tsirimonaki E, Marchetti B, et al. Down-regulation of MHC class I by bovine papillomavirus E5 oncoproteins Oncogene 2002; 21(2): 248-59.-52 Brady CS, Bartholomew JS, Burt DJ, et al. Multiple mechanisms underlie HLA dysregulation in cervical cancer Tissue Antigens 2000; 55(5): 401-11.].
Antigen processing consists of cellular and viral antigens being degraded by the proteasome. Resulting peptides are transported into the endoplasmatic reticulum by TAP, where they are loaded on empty MHC molecules. Complete MHC-peptide-complexes are then transported to the cell surface via the Golgi apparatus, and the peptides presented by MHC to the immune system.
Disruption of antigen processing and presentation mediated by HPV includes decreased expression of the proteasome subunits LMP2 and LMP7 [53 Evans M, Borysiewicz LK, Evans AS, et al. Antigen processing defects
in cervical carcinomas limit the presentation of a CTL epitope from
human papillomavirus 16 E6 J Immunol 2001; 167(9): 5420-8.], decreased expression of transporter subunits (TAP1 and TAP2) [53 Evans M, Borysiewicz LK, Evans AS, et al. Antigen processing defects
in cervical carcinomas limit the presentation of a CTL epitope from
human papillomavirus 16 E6 J Immunol 2001; 167(9): 5420-8.], and decreased expression of MHC-I itself [53 Evans M, Borysiewicz LK, Evans AS, et al. Antigen processing defects
in cervical carcinomas limit the presentation of a CTL epitope from
human papillomavirus 16 E6 J Immunol 2001; 167(9): 5420-8.] (Fig. 1). The E7 protein of high-risk HPV has been found to repress the MHC class I heavy chain gene promoter [54 Georgopoulos NT, Proffitt JL, Blair GE. Transcriptional regulation of
the major histocompatibility complex (MHC) class I heavy chain,
TAP1 and LMP2 genes by the human papillomavirus (HPV) type 6b,
16 and 18 E7 oncoproteins Oncogene 2000; 19(42): 4930-5.]. In addition, E7 also possesses the ability to repress the bi-directional promoter that regulates expression of both LMP2 and TAP1 [54 Georgopoulos NT, Proffitt JL, Blair GE. Transcriptional regulation of
the major histocompatibility complex (MHC) class I heavy chain,
TAP1 and LMP2 genes by the human papillomavirus (HPV) type 6b,
16 and 18 E7 oncoproteins Oncogene 2000; 19(42): 4930-5.]. The reduced expression of these antigen processing machinery components may cause impaired production of HPV epitopes, leading to a reduced HPV epitope repertoire [55 Riemer AB, Keskin DB, Zhang G, et al. A conserved E7-derived
cytotoxic T lymphocyte epitope expressed on human papillomavirus
16-transformed HLA-A2+ epithelial cancers J Biol Chem 2010;
285(38): 29608-2.]. HPV E5 has been reported to down-regulate HLA-A and -B cell surface expression, but no decrease was found in total HLA-C and -E class I expression [30 Venuti A, Paolini F, Nasir L, et al. Papillomavirus E5: the smallest
oncoprotein with many functions Mol Cancer 2011; 10: 140., 56 Ashrafi GH, Brown DR, Fife KH, Campo MS. Down-regulation of
MHC class I is a property common to papillomavirus E5 proteins Virus
Res 2006; 120(1-2): 208-11.]. This reduction is likely due to impaired MHC class I trafficking, caused by direct interaction of the E5 protein with the MHC class I heavy chain via the leucine pairs present in the first transmembrane domain [57 Ashrafi GH, Haghshenas MR, Marchetti B, O'Brien PM, Campo MS.
E5 protein of human papillomavirus type 16 selectively downregulates
surface HLA class I Int J Cancer 2005; 113(2): 276-83.]. Another mechanism is the arrest of MHC class I molecules in the Golgi apparatus (GA), caused by E5-induced alkalization of the GA and endosomes via interaction with the 16 kDa pore subunit of vacuolar-ATPase (V-ATPase) [58 Schapiro F, Sparkowski J, Adduci A, Suprynowicz F, Schlegel R,
Grinstein S. Golgi alkalinization by the papillomavirus E5 oncoprotein
J Cell Biol 2000; 148(2): 305-15., 59 Campo MS, Graham SV, Cortese MS, et al. HPV-16 E5 down-regulates
expression of surface HLA class I and reduces recognition by
CD8 T cells Virology 2010; 407(1): 137-42.]. Consistent with its role in the alkalinization of endosomes, E5 can prevent the endosomal breakdown of the invariant chain, a chaperone important in the maturation of MHC class II, leading to inhibition of expression of surface MHC class II [60 Zhang B, Li P, Wang E, et al. The E5 protein of human papillomavirus
type 16 perturbs MHC class II antigen maturation in human foreskin
keratinocytes treated with interferon-gamma Virology 2003; 310(1): 100-8.].
The functional effect of the down-regulation of MHC class I and class II molecules on the surface of HPV-positive cells is reduced recognition by T cells, leading to an escape from immunosurveillance. As HLA-C and -E are still expressed, HPV-infected cells do not become targets of natural killer (NK) cells, which recognize and kill cells with reduced MHC class I expression.
Modulation and Trafficking of Antigen Presenting Cells
Langerhans cells (LCs) are immature dendritic cells of myeloid origin resident in squamous epithelia. The normal function of LCs is to screen cell surfaces for pathogens, capture antigens by micropinocytosis or mannose receptor-mediated uptake, process captured proteins into immunogenic peptides, migrate out of the epithelium to the lymph nodes and present peptides in the context of MHC molecules to T cells, thereby initiating antigen-specific immune responses. Because of their role in initiating antiviral immune responses, DCs and LCs represent ideal targets for immune evasion by viruses.
It has been reported that upon HPV infection the number of LCs is significantly reduced in the infected section of the epithelium. This was attributed to E6-mediated reduced expression of the epithelial adhesion molecule E-cadherin on HPV-infected keratinocytes [61 Matthews K, Leong CM, Baxter L, et al. Depletion of Langerhans cells
in human papillomavirus type 16-infected skin is associated with E6-
mediated down regulation of E-cadherin J Virol 2003; 77(15): 8378-5., 62 D'Costa ZJ, Leong CM, Shields J, Matthews C, Hibma MH. Screening
of drugs to counteract human papillomavirus 16 E6 repression of E-cadherin
expression Invest New Drugs 2012; 30(6): 2236-51.]. LCs normally adhere to keratinocytes via this molecule, and migrate out of areas with reduced E-cadherin expression. Furthermore, E6 and E7 down-regulate macrophage inflammatory protein (MIP)-3α expression [63 Guess JC, McCance DJ. Decreased migration of Langerhans precursor-like
cells in response to human keratinocytes expressing human
papillomavirus type 16 E6/E7 is related to reduced macrophage
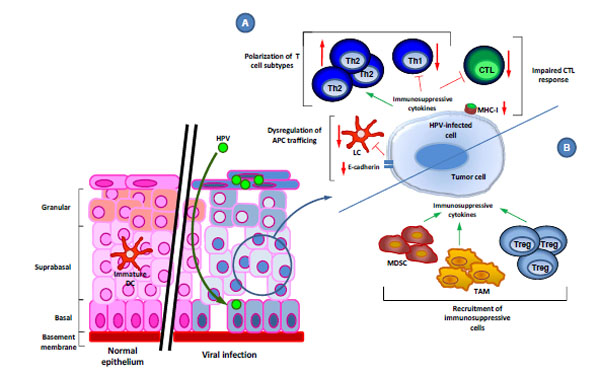
inflammatory protein-3alpha production J Virol 2005; 79(23): 14852-62.], leading to reduced attraction of APCs to the infected regions (Fig. 2 ).
).
The influence of HPV on the trafficking of immune cells has been investigated by employing HPV virus-like particle (VLP) technology. It has been found that the L1 major capsid protein, when self-assembled into VLPs, induces DC maturation through the overexpression of CXC receptor 4 (CXCR4), via the activation of the NF-κB and mitogen-activated protein kinase (MAPK) signaling pathways [64 Herman L, Hubert P, Herfs M, et al. The L1 major capsid protein of HPV16 differentially modulates APC trafficking according to the vaccination or natural infection context Eur J Immunol 2010; 40(11): 3075-84.]. L1 VLPs also increased the production of pro-inflammatory cytokines and chemokines by DCs, the enhanced migration of DCs, and the induction of an HPV16-specific CD8+ T cell response. However, natural HPV capsids are composed of L1 and L2, and HPV L1L2 VLPs do not induce LC maturation. Instead, they suppress the generation of an effective HPV-specific immune response via the deregulation of the PI3K-Akt pathway in LCs, suggesting that the HPV minor capsid protein is also involved in immune evasion, by inhibiting the maturation of LCs [65 Fausch SC, Fahey LM, Da Silva DM, Kast WM. Human papillomavirus can escape immune recognition through Langerhans cell phosphoinositide 3-kinase activation J Immunol 2005; 174(11): 7172-8., 66 Fahey LM, Raff AB, Da Silva DM, Kast WM. A major role for the minor capsid protein of human papillomavirus type 16 in immune escape J Immunol 2009; 183(10): 6151-.]. Taken together, these mechanisms result in suboptimal antigen capture and/or LC activation, which would be necessary for the initiation of anti-viral T cell responses.
Polarization of T Cell Phenotypes
The polarization of CD4+ T cells occurs upon interaction of naïve CD4+ T cells with their cognate antigen and depends on the cytokine environment. CD4+ T cells include T helper type 1 (Th1) cells, which stimulate cellular immune responses; Th2 cells, which promote humoral responses; Th17 cells, which contribute to the elimination of extracellular pathogens; and Foxp3+ regulatory T (Treg) cells, which prevent the development of autoimmunity. Th1 cells secrete high levels of IFN-γ and interleukin (IL)-2. The differentiation of Th1 is controlled by IL-12, in synergy with IL-18. IL-12 signals through JAK2 and TYK2, and activates mainly STAT4, a key transcription factor for Th1 consolidation [67 O'Shea JJ, Lahesmaa R, Vahedi G, Laurence A, Kanno Y. Genomic views of STAT function in CD4+ T helper cell differentiation Nat Rev Immunol 2011; 11(4): 239-50.]. Th2 cells secrete high amounts of IL-4, IL-5, IL-9 and IL-13. The acquisition of the Th2 phenotype is essentially driven by IL-4, which activates both JAK1 and JAK3 and the transcription factor STAT6 [68 Knosp CA, Johnston JA. Regulation of CD4+ T-cell polarization by suppressor of cytokine signalling proteins Immunology 2012; 135(2): 101-1.]. Several lines of evidence suggest that cell-mediated immune responses involving Th1 and CTL are essential for eradicating established HPV infections [69 Welters MJ, de Jong A, van den Eeden SJ, et al. Frequent display of human papillomavirus type 16 E6-specific memory t-Helper cells in the healthy population as witness of previous viral encounter Cancer Res 2003; 63(3): 636-41.-71 Bais AG, Beckmann I, Ewing PC, et al. Cytokine release in HR-HPV(+) women without and with cervical dysplasia (CIN II and III) or carcinoma, compared with HR-HPV(-) controls Mediators Inflamm 2007; 2007: 24147.].
A further immune evasion mechanism employed by HPV is to inhibit the Th1-response by inducing a shift from Th1 to Th2 (Fig. 2). HPV-related lesions have been found to be characterized by weak or absent IFN-γ-associated Th1 cell responses, but an up-regulation of Th2 cytokines (including IL-10) [72 Mota F, Rayment N, Chong S, Singer A, Chain B. The antigen-presenting
environment in normal and human papillomavirus (HPV)-related premalignant cervical epithelium Clin Exp Immunol 1999;
116(1): 33-40., 73 de Jong A, van Poelgeest MI, van der Hulst JM, et al. Human
papillomavirus type 16-positive cervical cancer is associated with
impaired CD4+ T-cell immunity against early antigens E2 and E6
Cancer Res 2004; 64(15): 5449-5.]. IL-10 production at an early HPV-associated disease stage may decrease immune recognition of HPV by down-regulating expression of HLA class I [72 Mota F, Rayment N, Chong S, Singer A, Chain B. The antigen-presenting
environment in normal and human papillomavirus (HPV)-related premalignant cervical epithelium Clin Exp Immunol 1999;
116(1): 33-40.] and up-regulation of non-classical HLA-G molecules [74 Rodriguez JA, Galeano L, Palacios DM, et al. Altered HLA class I and
HLA-G expression is associated with IL-10 expression in patients with
cervical cancer Pathobiology 2012; 79(2): 72-83.]. It has been demonstrated that HLA-G can inhibit the activity of CTLs as well as natural killer (NK) cells [75 Le Gal FA, Riteau B, Sedlik C, et al. HLA-G-mediated inhibition of
antigen-specific cytotoxic T lymphocytes Int Immunol 1999; 11(8): 1351-6., 76 Marchal-Bras-Goncalves R, Rouas-Freiss N, Connan F, et al. A soluble
HLA-G protein that inhibits natural killer cell-mediated cytotoxicity
Transplant Proc 2001; 33(3): 2355-9.]. Moreover, HLA-G may modulate DC maturation, trafficking, antigen presentation and their cross-talk with T and NK cells [77 Gros F, Cabillic F, Toutirais O, Maux AL, Sebti Y, Amiot L. Soluble
HLA-G molecules impair natural killer/dendritic cell crosstalk via
inhibition of dendritic cells Eur J Immunol 2008; 38(3): 742-9.].
Along the same lines, CD1d expression is changed in HPV-infected cells. CD1d is an important player in innate immune responses and can modulate adaptive immune cells by altering the Th1/Th2 polarization. The E5 protein interacts with the chaperone calnexin in the endoplasmic reticulum (ER) and likely impairs calnexin-mediated CD1d folding, thereby arresting CD1d molecules in the ER and interrupting appropriate trafficking of CD1d to the surface of HPV-infected cells. It has also been demonstrated that E5 inhibits CD1d-mediated IL-12 production [78 Miura S, Kawana K, Schust DJ, et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: a possible mechanism for immune evasion by HPV J Virol 2010; 84(22): 11614-23.].
Taken together, HPV-infection leads to a polarization of the composition of effector T cells in the lesion microenvironment that favors immune escape.
IMMUNOSUPPRESSIVE MECHANISMS IN HPV-DRIVEN TUMORS
All immune evasion mechanisms discussed so far were studied relating to productive viral infection. Here, immune evasion increases the duration and size of HPV infection, thus raising the probability that some infected cells will become transformed [30 Venuti A, Paolini F, Nasir L, et al. Papillomavirus E5: the smallest oncoprotein with many functions Mol Cancer 2011; 10: 140.]. Of note, most studies were done in vitro, and utilized overexpressed HPV proteins or even HPV-transformed cell lines. Thus, these data only mimic HPV infection of normal human keratinocytes and still need to be confirmed in vivo.
Upon malignant transformation, the HPV genome is often integrated into the DNA of the host cell. This integration is a terminal event for the life cycle of the virus, as many genes are lost in this process. It is therefore important to differentiate between immune evasion mechanisms which are active in HPV infection, and immune evasion mechanisms employed by HPV-driven tumors.
Most of the discussed immune evasion mechanisms are active in both scenarios, but there are several differences. First, one immune evasion mechanism is lost in tumor cells. As explained above, during the productive phase of the HPV life cycle, episomal transcription is tightly regulated by the viral transcription factor E2, resulting in low-level expression of E6 and E7. E2 is lost upon integration, therefore E6 and E7 are no longer kept at low levels, but are highly expressed [3 zur Hausen H. Papillomaviruses in human cancer Appl Pathol 1987; 5(1): 19-24., 79 Romanczuk H, Howley PM. Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity Proc Natl Acad Sci U S A 1992; 89(7): 3159-63., 80 Jeon S, Lambert PF. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis Proc Natl Acad Sci U S A 1995; 92(5): 1654-8.]. This not only results in more targets for immune attack, but unfortunately also increases the risk of accumulation of cellular genetic and epigenetic changes and thus malignant transformation [81 Pett MR, Alazawi WO, Roberts I, et al. Acquisition of high-level chromosomal instability is associated with integration of human papillomavirus type 16 in cervical keratinocytes Cancer Res 2004; 64(4): 1359-68.].
Second, several immune evasion strategies exist exclusively in tumors, and not in normal HPV infection. These include the recruitment of immune cells with immunosuppressive properties (Fig. 2). Increasing evidence indicates that tumor-associated macrophages (TAMs) and other myeloid-derived cells such as myeloid-derived suppressor cells (MDSCs) play an important role in HPV-associated carcinogenesis. TAMs are macrophages differentiated predominantly into a M2 phenotype [82 Sica A, Larghi P, Mancino A, et al. Macrophage polarization in tumour
progression Semin Cancer Biol 2008; 18(5): 349-55., 83 Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated
macrophages and the related myeloid-derived suppressor
cells as a paradigm of the diversity of macrophage activation Hum
Immunol 2009; 70(5): 325-0.] and are able to suppress antiviral T cell responses [84 Lepique AP, Daghastanli KR, Cuccovia IM, Villa LL. HPV16 tumor
associated macrophages suppress antitumor T cell responses Clin
Cancer Res 2009; 15(13): 4391-00.]. These myeloid cells secrete regulatory cytokines such as IL-10 and transforming-growth factor β (TGFβ) and induce T regulatory cells or lymphocyte cell death [82 Sica A, Larghi P, Mancino A, et al. Macrophage polarization in tumour
progression Semin Cancer Biol 2008; 18(5): 349-55., 83 Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated
macrophages and the related myeloid-derived suppressor
cells as a paradigm of the diversity of macrophage activation Hum
Immunol 2009; 70(5): 325-0.]. Elevated levels of IL-10 as well as increased numbers of tumor infiltrating macrophages are correlated with progression of HPV-associated lesions [85 Clerici M, Merola M, Ferrario E, et al. Cytokine production patterns in
cervical intraepithelial neoplasia: association with human
papillomavirus infection J Natl Cancer Inst 1997; 89(3): 245-50., 86 Bolpetti A, Silva JS, Villa LL, Lepique AP. Interleukin-10 production
by tumor infiltrating macrophages plays a role in Human
Papillomavirus 16 tumor growth BMC Immunol 2010; 11: 27.]. Moreover, MDSCs are also characterized by significant production of reactive oxygen species (ROS) triggered by ARG1 [87 Bronte V, Serafini P, Mazzoni A, Segal DM, Zanovello P. L-arginine
metabolism in myeloid cells controls T-lymphocyte functions Trends
Immunol 2003; 24(6): 302-6.]. ROS may induce T cell dysfunction through the down-regulation of the CD3 ζ-chain of the T cell receptor complex [88 Otsuji M, Kimura Y, Aoe T, Okamoto Y, Saito T. Oxidative stress by
tumor-derived macrophages suppresses the expression of CD3 zeta
chain of T-cell receptor complex and antigen-specific T-cell responses
Proc Natl Acad Sci U S A 1996; 93(23): 13119-24.], thus MDSCs are able to inhibit CTL activation [89 Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific
inhibition of CD8+ T cell response by immature myeloid cells in cancer
is mediated by reactive oxygen species J Immunol 2004; 172(2): 989-9.]. However, the impact of MDSCs on HPV-associated lesions is not yet fully elucidated.
A large number of studies support a crucial role of CD4+ CD25+ regulatory T cells (Tregs) in immune homeostasis, tolerance, infection and tumor immunity [90 Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells Nat Immunol 2003; 4(4): 330-6., 91 von Boehmer H. Mechanisms of suppression by suppressor T cells Nat Immunol 2005; 6(4): 338-44.]. Tregs have been observed to infiltrate tumor masses, especially in the early stage of tumor progression [92 Piersma SJ, Welters MJ, van der Burg SH. Tumor-specific regulatory T cells in cancer patients Hum Immunol 2008; 69(4-5): 241-9.]. Tregs are able to strongly inhibit cytokine production and proliferation of activated naive CD4+ T cells and natural killer cells (NK) as well as prevent activation of CTLs. Additionally, Tregs can activate immunosuppressive functions of immune cells by IL-10 as well as TGF β-dependent mechanisms [91 von Boehmer H. Mechanisms of suppression by suppressor T cells Nat Immunol 2005; 6(4): 338-44., 92 Piersma SJ, Welters MJ, van der Burg SH. Tumor-specific regulatory T cells in cancer patients Hum Immunol 2008; 69(4-5): 241-9.]. Increased frequencies and suppressive activity of Tregs have been observed in cervical cancer patients, both at the tumor site and in draining lymph nodes, and are likely to contribute to the impaired HPV-specific immune response observed in these patients [92 Piersma SJ, Welters MJ, van der Burg SH. Tumor-specific regulatory T cells in cancer patients Hum Immunol 2008; 69(4-5): 241-9., 93 Visser J, Nijman HW, Hoogenboom BN, et al. Frequencies and role of regulatory T cells in patients with (pre)malignant cervical neoplasia Clin Exp Immunol 2007; 150(2): 199-209.]. Increased Tregs numbers have been found to be strongly associated with an increased risk for progression of premalignant lesions to cancer [94 Molling JW, de Gruijl TD, Glim J, et al. CD4(+)CD25hi regulatory T-cell frequency correlates with persistence of human papillomavirus type 16 and T helper cell responses in patients with cervical intraepithelial neoplasia Int J Cancer 2007; 121(8): 1749-55., 95 Adurthi S, Krishna S, Mukherjee G, Bafna UD, Devi U, Jayshree RS. Regulatory T cells in a spectrum of HPV-induced cervical lesions: cervicitis, cervical intraepithelial neoplasia and squamous cell carcinoma Am J Reprod Immunol 2008; 60(1): 55-65.]. However, it is still unclear whether persistent HPV infection is responsible for increased numbers of Tregs, or whether increased Treg frequencies lead to persistence.
FINAL REMARKS
HPV has evolved to evade human immune detection in multiple ways to establish an infection and maintain a persistent life cycle that leads to viral reproduction. Exactly this persistence is the greatest risk factor for the development of HPV-mediated invasive malignancies.
The most important HPV immune evasion mechanism is to become invisible to the host immune system. This includes the absent triggering of any danger signals, as HPV infection causes no cytolysis, no cytopathic cell death, and no inflammation. Suppression of the interferon response, resistance to immune-mediated apoptosis, down-regulation of adhesion molecules for APCs, and active MHC class I down-regulation and impaired antigen presentation have also been shown to play a critical role in HPV immune evasion.
Nevertheless, most HPV-associated lesions are eventually eradicated by the host immune system, and only a small percentage progresses to invasive malignancies. Virus-specific CD4+ and CD8+ T cell responses are essential for the immune control of HPV infection. Therefore, therapeutic vaccines eliciting the desired immune responses are considered to be attractive treatment options, and are actively investigated. Unfortunately, most HPV immunotherapies studies to date have yielded disappointing clinical results. A detailed understanding of the molecular mechanisms underlying HPV immune escape is therefore necessary to further improve therapeutic vaccine strategies.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.