- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




The Open Virology Journal
(Discontinued)
ISSN: 1874-3579 ― Volume 15, 2021
An Influenza Virus M2 Protein Specific Chimeric Antigen Receptor Modulates Influenza A/WSN/33 H1N1 Infection In Vivo
Simon J Talbot*, 1, Natalie F Blair 1, Niolette McGill 1, Yvonne Ligertwood 2, Bernadette M Dutia 2, Ingo Johannessen 1
Abstract
A potential target for the development of universal vaccine strategies against Influenza A is the M2 protein – a membrane protein with a highly conserved extracellular domain. In this study we developed engineered T-cell receptors, by fusing M2-specific antibody sequences with T-cell receptor transmembrane and signaling domains to target influenza infected cells. When expressed on T-cells, these novel T-cell receptors (chimeric antigen receptors - CARs) are able to recognize specific antigens on the surface of target cells via an MHC-independent mechanism. Using an existing monoclonal antibody (14C2) specific for the M2 ectodomain (M2e), we generated an M2-specific CAR. We tested the specificity of this M2 CAR in vitro by measuring the activation of T-cells in response to M2-specific peptides or M2-expressing cell lines. Both Jurkat T-cells and peripheral blood mononuclear cells expressing the M2-specific CAR responded to specific antigen stimulation by upregulating NFAT and producing γ-interferon. To test whether the M2-specific CAR are effective at recognizing influenza infected cells in vivo we used an established BALB/c murine infection model. At day 4 post-infection, when M2 CAR expressing splenocytes could be detected in the lung, the Influenza A/WSN/33 virus titre was around 50% of that in control mice. Although the lung virus titre later increased in the treated group, virus was cleared in both groups of mice by day 8. The results provide support for the development of M2e as a target for cell mediated immunotherapy.
Article Information
Identifiers and Pagination:
Year: 2013Volume: 7
First Page: 28
Last Page: 36
Publisher Id: TOVJ-7-28
DOI: 10.2174/1874357901307010028
Article History:
Received Date: 15/11/2012Revision Received Date: 7/1/2013
Acceptance Date: 11/1/2013
Electronic publication date: 25/2/2013
Collection year: 2013
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the University of Edinburgh, Division of Pathway Medicine, Chancellors building, Little France, Edinburgh EH16 4SB, UK; Tel: +44 131 2426282; E-mail: stalbot@ed.ac.uk
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 15-11-2012 |
Original Manuscript | An Influenza Virus M2 Protein Specific Chimeric Antigen Receptor Modulates Influenza A/WSN/33 H1N1 Infection In Vivo | |
INTRODUCTION
Influenza A virus poses a continued threat to the world’s population as a result of its pandemic potential and the risk of severe influenza A-induced disease with the emergence of a pandemic strain. The limited range of antiviral drugs and concerns about their effectiveness underscore the importance of developing novel treatment.
A member of the orthomyxoviridae family, influenza A virus genome is comprised of 8 single-stranded negative-sense RNA segments with an overall size of approximately 14kb [1Lamb RA, Krug RM. Orthomyxoviridae: The viruses and their replication In: Knipe DM, Howley PM, Eds. Fields virology. 4th edition. Philadelphia: Lippincott, Williams & Wilkins Publishers 2001; pp. 1487-531.]. The virus has an avian reservoir and the potential to cause pandemics due to its ability to undergo antigenic shift (re-assortment of genetic segments in co-infected cells). Influenza A-derived proteins include the surface antigens haemagglutinin (H; receptor binding protein) and neuraminidase (N; fusion of virus envelope with target cell membrane) that are highly variable and define virus strains [2Wong SSY, Yuen K-Y. Avian Influenza Virus Infections in Humans Chest 2006; 129: 156-68.]. There are 17 (H1-17) and 9 (N1-9) recognised subtypes of the H and N antigens, respectively, and antigenic variability results in influenza A epidemics. The N antigens have sialidase function that removes sialic acid from glycosilated molecules thereby facilitating release of virus during egress from host cell by cleaving glycosidic linkages to sialic acid on virion and host cell surfaces. Neuraminidase inhibitors (NIs; oseltamivir, zanamivir) block this stage of infection [3von Itzstein M, Wu W-Y, Kok GB, et al. Rational Design of Potent Sialidase-Based Inhibitors of Influenza Virus Replication Nature 1993; 363: 418-23., 4Moscona A. Neuraminidase Inhibitors for Influenza N Engl J Med 2005; 353: 1363-73.]. The highly conserved matrix (M) 1 and M2 proteins form a shell around nucleic material and an ion channel in the viral envelope, respectively [5Pinto LH, Lamb RA. The M2 Proton Channels of Influenza A and B Viruses J Biol Chem 2006; 281(14): 8997-9000.]. The M2 protein, an abundantly expressed integral membrane protein of 97 amino acids has an extracellular domain composed of 23 non-glycosylated N-terminal residues, the M2 ectodomain (M2e). M2e is highly conserved across influenza A virus. Whilst resistance against the M2 ion channel inhibitors (amantadine and rimantadine) is widespread amongst avian influenza [6Beigel JH, Farrar J, Han AM, et al. Avian Influenza A (H5N1) Infection in Humans N Engl J Med 2005; 353: 1374-85.], such resistance is not associated with M2e.
The immune response to influenza A virus infection includes long-lived (serum and nasal secretory) antibody responses that are subtype-specific for the infecting virus. Neutralising antibodies against H antigens confer resistance to infection although re-infection with the same virus can occur [7Lewis DB. Avian Flu to Human Influenza Ann Rev Med 2006; 57: 139-54.]. Antibodies against the N antigens reduce disease severity and transmission risk. Antibodies also arise against the M proteins although M2e antibodies are only present in low titres. M2e induces a poor antibody response in humans, but its conserved nature across influenza A strains and the ability of M2e-specific antibodies to restrict virus replication in vivo has made it a strong vaccine candidate [8Gerhard W, Mozdzanowska K, Zharikova D. Prospects for Universal Influenza Virus Vaccine Emerg Infect Dis 2006; 12(4): 569-74.]. Infection also results in cellular immunity mediated by cytotoxic T lymphocytes (CTLs) which lyse virus-infected cells. Both H and N antigens stimulate CTLs which are key players in eradicating and curtailing infection [9Doherty PC, Turner SJ, Webby RG, Thomas PG. Influenza and the Challenge for Immunology Nature Immun 2006; 7(5): 449-55.].
Avian influenza A gave rise to all known human pandemics in the 20th century, and the first pandemic of the 21st century (‘swine flu’) was a composite of avian, swine and human strains that emerged from pigs in Mexico in 2009. The global population continues to be threatened by a new influenza A virus pandemic - and the most likely agent is the H5N1 virus. Since its resurgence in 2003, avian influenza H5N1 has caused 607 confirmed human cases in Asia, the Middle East and Europe (Turkey); to date, 358 (59%) individuals have succumbed to the infection [10World Health Organization. ‘Cumulative Number of Confirmed Human Cases of Avian Influenza A/(H5N1) Reported to WHO’ http://www.who.int/influenza/human_animal_interface/EN_GIP_20120706CumulativeNumberH5N1cases.pdf [ACCESSED 12 July 2012];]. Whilst H5N1 has all the attributes of a pandemic strain in birds (ie, it is a new and virulent strain that transmits between birds), it still does not spread efficiently between humans. But that may change if the virus adapts to its human host. Antiviral therapy is the mainstay of control of H5N1 influenza infection and antiviral drugs (primarily, the NIs) play a key role in current avian influenza preparedness plans. The NIs are effective against influenza A if administered within 48 hours from onset of clinical symptoms/signs and work by inhibiting N antigen-mediated virus egress - but avian influenza may need high doses and prolonged treatment (‘relative resistance’) which compounds the supply of these agents [11de Jong MD, Tran TT, Truong HK, et al. Oseltamivir Resistance During Treatment of Influenza A (H5N1) Infection N Engl J Med 2005; 353: 2667-72.]. Furthermore, in light of concerns about their effectiveness against H5N1 avian influenza [12Mai Le Q, Kiso M, Someya K, et al. Avian flu. isolation of drug-resistant H5N1 virus (brief communication) Nature 2005; 437: 1108., 13McCullers JA. The Clinical Need for New Antiviral Drugs Directed against Influenza Virus J Infect Dis 2006; 193: 751-3.] and evidence of possible resistance to the drugs, it is of paramount importance to develop novel treatment. One such approach involves adoptive immunotherapy, and chimeric antigen receptor (CAR) technology is one such option.
Genetically engineered T cells offer an exciting alternative to CTLs. The aim of CAR technology is to modify the T cell receptor (TCR) in a manner that replaces the antigen recognition part with a single chain antibody-like molecule thereby circumventing MHC-restriction and re-directing CTLs to novel surface targets [14Schumacher TNM. T-Cell-Receptor Gene Therapy Nat Rev Immun 2002; 2: 512-9.]. The vectors may also contain additional molecular machinery (for example, lymphocyte triggering receptors TCR-ζ and FcR-γ) to enhance homing, cytotoxicity and survival of the transfected CTLs to create novel CARs (‘T bodies’). TCRs specific for viral antigens have been used previously to re-direct T cells against virus-infected cells as well as in non-viral-associated cancer where a tumour-specific surface marker is available [15Orentas RJ, Roskopf SJ, Nolan GP, et al. Retroviral Transduction of a T-Cell Receptor Specific for an Epstein-Barr-Virus-Encoded Peptide Clin Immunol 2001; 96: 220-8., 16Haynes NM, Trapani JA, Teng MWL, et al. Single-Chain Antigen Recognition Receptors that Costimulate Potent Rejection of Established Experimental Tumours Blood 2002; 100: 3155-63.]. The conserved region of the M2e protein is exposed on the surface of influenza A virus-infected cells and is, thus, a potential target for CAR T cells.
Originally described by Zebedee and Lamb in 1988, the 14C2 monoclonal antibody (MAb) recognizes the M2e protein [17Zebedee SL, Lamb RA. Influenza A Virus M2 Protein: Monoclonal Antibody Restriction of Virus Growth and Detection of M2 in Virions J Virol 1988; 62(8): 2762-72.] and is broadly reactive against several Influenza A subtypes. This study delineated the 14C2 antigen recognition sequences (paratopes) of the 14C2 hybridoma cell line and used the information to develop 14C2 CAR T cells that were re-directed to influenza A virus-infected cells. Using this novel approach in in vitro and in vivo systems, the project generated translational data which will not only be applicable to influenza A, but also to other virus infections.
MATERIALS AND METHODS
PBMCs
Anonymised buffy coat donations were provided by the Scottish National Blood Transfusion Service (SNBTS) from registered blood donors with informed consent. Heparin (1,000 IU into 50 ml; CP Pharmaceuticals, Wrexham, UK) was added to buffy coat samples, and peripheral blood mononuclear cells (PBMCs) were separated using Histopaque-1077 (Sigma, Irvine, UK). PBMCs were washed in RPMI 1640 (Invitrogen), and then cryopreserved in 10% dimethyl sulphoxide (Sigma) in foetal bovine serum (FBS; Invitrogen).
Cell Lines and Virus
293T/Phoenix Amp retroviral packaging cells (obtained from Prof. R Debets; University of Rotterdam), Madin–Darby canine kidney epithelial cells (MDCK; ATCC), and HT1080 cells (ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% FBS (Invitrogen), 100U/ml penicillin, 50 µg/ml streptomycin. The Lymphoblastoid cell line LCL114 was derived by in vitro EBV infection of PBMCs from healthy donors (Wilkie et al., 2004) and cultured in RPMI 1640 (Invitrogen) supplemented with 10% FBS (Invitrogen) and 2mM L-glutamine, 100U/ml penicillin, 50 µg /ml streptomycin. The A/WSN/33 (H1N1) influenza virus, obtained from Dr D. Jackson, University of St Andrews, UK, was propagated in MDCK cells prior to use in this study.
Construction of 14C2 CAR
The 14C2 Hybridoma cell line was obtained from Prof Robert Lamb (University of Illinois) and cultured in RPMI 1640 supplemented with 1 mM pyruvate, 2mM L-glutamine, hybridoma enhancing supplement (Sigma) and 20% FBS (Invtrogen). Total RNA was extracted from 1×106 hybridoma cells using the RNeasy kit from Qiagen and treated with DNAse (RNAse deficient - Promega) to remove any contaminating DNA. 1 µg of DNase treated RNA was converted to cDNA using oligo dT primers and MMLV reverse transcriptase (Affinity Script Multiple Temperature cDNA synthesis kit, Stratagene). Using a mouse IgG primer set (Calbiochem) the variable heavy and light chain cDNA sequences were amplified and cloned into the PGEM-T easy vector (Promega). Clones were analysed by sequencing to confirm the presence of murine IgG sequences. Variable heavy and variable light chain sequences were joined with a Serine/Glycine linker to form a ScFv using PCR. The VH was amplified using the primers VH forward; 5’- ACTGCCATGGCCCAGGTCCAGCTGCAGCAG-‘3 and VH reverse; 5’ GCCGCTGCCACCTCCGCCTGAACCGCCTCCACCGCTCGAGACAGTGACCAGAGTCCC- ‘3. The VL was amplified using VL forward; 5’-TCAGGCGGAGGTGGCAGCGGCGGTGGCGGGTCGACGGACATTGTGATGACACAGTC-3 and VL reverse; 5’-TTTCTCGTGCGGCCGCACGTTTCAGCTCCAACTTGG 3’. Both PCR products were purified from a 2% agarose gel, 50 µg of each mixed together, annealed at 65°C, and subject to 5 rounds of PCR before the addition of the VH forward primer and the VL reverse primer to amplify the full length product (Fig. 1A ). The 14C2 ScFv PCR product was cloned into the retroviral vector PMP71.tCD34.2A.CD19.CD3zeta [18Cheadle EJ, Hawkins RE, Batha H, O'Neill AL, Dovedi SJ, Gilham DE. Natural Expression of the CD19 Antigen Impacts the Long-Term Engraftment but Not Antitumor Activity of CD19-Specific Engineered T Cells J Immunol 2010; 184: 1885-96.] (Fig. 1B) by replacing the CD19 encoding sequence by digestion with ClaI and NotI.
). The 14C2 ScFv PCR product was cloned into the retroviral vector PMP71.tCD34.2A.CD19.CD3zeta [18Cheadle EJ, Hawkins RE, Batha H, O'Neill AL, Dovedi SJ, Gilham DE. Natural Expression of the CD19 Antigen Impacts the Long-Term Engraftment but Not Antitumor Activity of CD19-Specific Engineered T Cells J Immunol 2010; 184: 1885-96.] (Fig. 1B) by replacing the CD19 encoding sequence by digestion with ClaI and NotI.

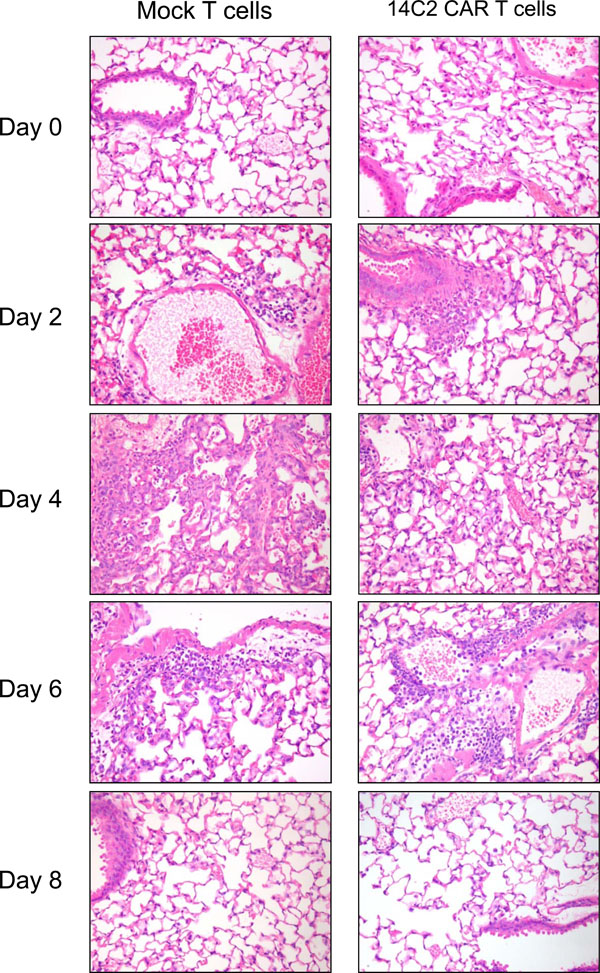 |
Fig. (6) H&E staining of lung sections taken at day 0, 2, 4, 6 and 8 post infection. Significant inflammation and lymphocyte infiltration is observed on days 2, 4, and 6 pi. |
Peptides
All peptides were synthesised by Thistle Scientific (Scotland). Three M2 specific peptides were used:
M2 terminal peptide; SLLTEVETP,
M2e; SLLTEVETPIRNEWGCRCNDSSD, and
M2 tetramer; SLLTEVETPIRNEWGCRCNDSSDGGG]4-(branched K).
An unrelated peptide LIALWNLHGQALFLG (LMP-1) was used as a negative control.
Transduction of Human PBMCs
The packaging cells (293T/Phoenix-Amp) were seeded at 1.5×106 cells per 75 cm2 flask coated with 0.1% gelatin/PBS and grown overnight at 37°C in 10 ml DMEM (10% FBS, 100U/ml penicillin, 50 µg/ml streptomycin, 2mM L-Glutamine). The following day, the media was refreshed 3 hours prior to transfection of the cells. PBMCs (from frozen stock) were resuspended in RPMI (20% FBS, 100U/ml penicillin, 50 µg/ml streptomycin, 2mM L-Glutamine) at a cell density of 2×106/ml, and stimulated with OKT3 (anti-CD3 MAb) to a final concentration of 10 ng/ml, for 2 days (37°C / 5% CO2). The packaging cell line was transfected with the 14C2 retroviral CAR construct and helper constructs pHIT60 and pColtGalV (provided by Prof R Debets, University of Rotterdam) (10 µg of each) to a total amount of 40 µg DNA, using the Calcium Phosphate transfection kit (Promega). Cells were incubated for 2 days (37°C/5% CO2). Retrovirus was harvested from the transfected packaging cells and used to infect activated PBMCs. The virus supernatant was filtered through a 0.45 µm filter and IL-2 added to a final concentration of 100 IU/ml. 2 mls of virus was added to each well of a Non-tissue culture 24-well plate previously coated with retronectin (12 µg/ml in dH2O; Takara, Cambrex), and centrifuged for 1 hour at 1000×g. Following centrifugation the supernatant was removed and 1×106 activated PBMCs were then added per well. The cells were then centrifuged for 1 hour at 1000×g and incubated 4-5 hours (37°C/5% CO2). The media was aspirated carefully from the wells and followed by addition of 2 ml RPMI (20% FBS, 100U/ml penicillin, 50 µg/ml streptomycin, 2mM L-Glutamine, 100 IU/ml IL-2) and incubated overnight (37°C/5% CO2). Transduction efficiency was assessed at day 4 post transduction, using a CD34 specific antibody (Becton Dickinson; clone 582) to measure the expression of tCD34 on the surface of transduced cells using flow cytometry.
Transduction of Murine Splenocytes
The packaging cells (293T/Phoenix Amp) were prepared and transfected as above except that the pColtGalV plasmid was replaced with pHIT123 (MLV-E Env, provided by Prof. R. Debets, University of Rotterdam). Spleens were isolated from Balb/c mice and passed through a 70 μm cell strainer into a 50 ml tube using 20mls of PBS. The harvested splenocytes were centrifuged, resuspended in 5 ml of filtered erylysis buffer (155 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.4) and incubated for 4 min at 4°C. The cells were then washed twice in 20 ml of RPMI 1640 medium, before resuspending in 30 ml RPMI 1640 medium (+10% FBS, 100U/ml penicillin, 50 µg/ml streptomycin, 2mM L-Glutamine, 1 mM NaPyruvate, 50 μM β-mercaptoethanol) and counted. Splenocytes were seeded at 5x106/ml in a T25 flask before addition of 2.5 μg/ml Concanavalin A (Calbiochem), 1ng/ml IL-7 (Santa Cruz) and 100 U/ml human rIL-2, and incubated for 24 hours. The activated splenocytes were then transduced with retrovirus harvested from the packaging cells using retronectin coated plates as described above. The transduced splenocytes were cultured in fresh culture media with 100 U/ml IL-2, at a cell density of 0.15×106/ml in 2 ml per well of a 24 well plate for 2 days before being assessed for transduction efficiency using flow cytometry and a CD34 antibody.
Flow Cytometry
Cells to be analysed were washed twice in PBSA (1% BSA, 0.1% sodium azide, 0.2% EDTA) by centrifugation at 120×g for 5 minutes. The supernatant was decanted and cells resuspended in residual liquid. 10µL of the anti-CD34 (clone 582) antibody was added and the tubes incubated for 20 minutes at 4°C in the dark. Cells were washed twice in PBSA then resuspended in 1x CellFix (Becton Dickinson) and evaluated on a FACScan (Becton Dickinson) flow cytometer. At least 10,000 events were acquired for each sample. Data were subsequently analysed using the CellQuest software.
M2 Expressing Cell Lines
The following oligonucleotides were annealed and subject to PCR to generate a dsDNA encoding the Influenza M2 protein with a deletion of residues 29-31 and 62:
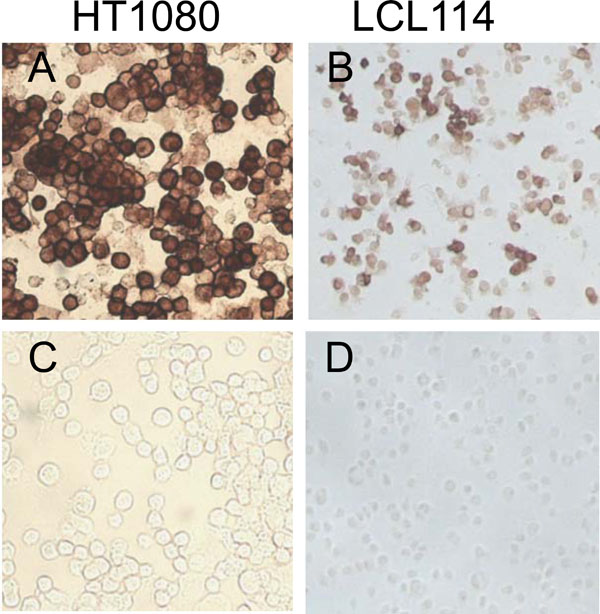
5’CGGGATCCATGAGTCTTCTAACCGAGGTCGAAACGCCTATCAGAAACGAATGGGGGTGCAGATGCAACGATTCAAGTGATCCTCTCGTCATTATCATTGAGATCTTGC-3’ and 5’-TGCCTCGAGTAGTTTTTTATCTTTTC AAACCGTATTTAAAGCGACGATAAATGCATTTGAAAAAAAGACGATCAAGAATCCACAATATCAAGTGCAAGATCTCAATGATAATGACG-3’. The DNA was cloned into the retroviral expression vector pFB-Neo (Stratagene) using the restriction enzymes BamHI and XhoI. Recombinant retrovirus was produced by transfecting the pFB-Neo M2 plasmid into the packaging cells (293T/Phoenix Amp) as described for transduction of human PBMCs. The resulting retrovirus was then used to transduce the cell lines HT1080 and LCL114 (EBV transformed Lymphoblastoid cells line). Expression of the M2 protein was confirmed by staining with the 14C2 Monoclonal antibody (Fig. 2 ).
).
ELISPOT
The Elispot technique is an established assay used to determine the number of γ-IFN releasing cells. Using PBMCs transduced with the M2 specific CAR, we used the ELISPOT assay to determine the number of γ-IFN releasing cells in response to specific stimulation with M2 peptides or M2 expressing cell lines. Briefly, 0.2x106 14C2 CAR transduced PBMCs in 100µl of culture media were stimulated with 10µl peptide (100µg/ml) or 0.1x106 M2 expressing LCL114 for 24 hours, and assayed for the expression of gamma interferon according to manufacturers instructions (Mabtech).
T-Cell Activation Assay - NFAT Reporter Assay
5×106 Jurkat E6.1 T cells were transiently nucleofected with 5 μg GLuc-NFAT(RE)6 and 5 μg of 14C2 CAR construct using an Amaxa nucleofector (AmaxaBiosystems, Cologne, Germany) as described [19Govers C, Sebestyén Z, Berrevoets C, Venselaar H, Debets R. T Cell Receptor Fused to CD3ζ: Transmembrane Domain of CD3ζ Prevents TCR Mis-Pairing, Whereas Complete CD3ζ directs functional TCR expression Open Gene Ther J 2011; 4: 11-22.]. Briefly, T cells were resuspended in 100 μl supplemented buffer V to which was added and pulsed with the Nucleofector set at program C-16. Next, T cells were immediately transferred to 2.5 ml warm Jurkat T-cell medium in T25 flasks for overnight recovery (37°C, 5% CO2). 0.2 ×106 transfected Jurkat T-cells were transferred to each well of a 96 well plate and stimulated with peptides (1µg/ml) or 0.1×106 M2 expressing cell lines for 24 hours. Subsequently, 25 µl of cell supernatant was assayed for luciferase activity according to manufacturer`s instructions (Gluc substrate; New England Biolabs) using a Turner systems Luminometer.
Mouse Model of Influenza Virus Infection
BALB/c mice were purchased from Harlan UK Ltd (Oxon, UK). All work was carried out under a UK Home Office licence according to the Animals (Scientific Procedures) Act 1986. Ten-week-old female mice were used in all experiments. Mice were anaesthetized using halothane (Rhone Merieux Ltd) and 1×106 transduced murine splenocytes were transferred to each mouse via the tail vein. 24 hrs after the transfer of transduced splenocytes, the mice were anaesthetized and infected intranasally with 5×102 p.f.u. of A/WSN/33 influenza virus in 40 µl PBS volume. Mice were weighed daily and assessed for visual signs of clinical disease, including inactivity, ruffled fur and laboured breathing. At this virus dose, animals do not exhibit clinical signs or weight loss. At various times after infection, mice were euthanized by CO2 asphyxiation, and the lungs removed, homogenized in PBS and clarified by centrifugation. Titres of infectious virus were determined by standard plaque assays on MDCK cells.
tCD34 PCR
DNA was extracted from lung tissue using the DNeasy blood and tissue DNA kit, according to manufacturers instructions (Qiagen). 100µg of extracted DNA was amplified using a nested PCR protocol with tCD34 specific primers (1st round amplification: 5’-CAAGCCCTATACCTCCTCTT-3’ and 5’-GTGTTTTCTGGCTGTAGCTC-3’, 2nd round amplification: 5’-CTGCGCCGAGTTTAAGAAAG-3’ and 5’-TGCTCGGTAAAGTCCAGGAT-3’) or murine GAPDH primers (5’-GCCCATCACCATCTTCCAG-3’ and 5’-TGAGCCCTTCCACAATGCC-3’).
Statistical Analysis
Data were analysed using Graphpad Prism software (San Diego, CA). For the mouse infection experiments data were analysed using a two-way Anova with Bonferoni post-tests.
RESULTS
Generation of M2 Specific ScFv
The M2e-specific 14C2 mAb was generated using M2 protein purified from A/WSN/33-infected CV1 cell lysates [17Zebedee SL, Lamb RA. Influenza A Virus M2 Protein: Monoclonal Antibody Restriction of Virus Growth and Detection of M2 in Virions J Virol 1988; 62(8): 2762-72.]. The antibody-binding site was located to the M2 ectodomain (M2e). Using the Hybridoma cell line (Prof RA Lamb, Northwestern University, Illinois, USA), we amplified the MAb variable chain regions and joined them together with a Serine/Glycine spacer sequence using PCR. The sequence of the resulting Single chain antibody (ScFv) is shown in Fig. 1A.
Construction of M2 Specific Chimeric Antigen Receptor
The 14C2 ScFv sequence was cloned into a retroviral expression vector [18Cheadle EJ, Hawkins RE, Batha H, O'Neill AL, Dovedi SJ, Gilham DE. Natural Expression of the CD19 Antigen Impacts the Long-Term Engraftment but Not Antitumor Activity of CD19-Specific Engineered T Cells J Immunol 2010; 184: 1885-96.]. This construct (Fig. 1B) places the ScFv sequence in frame with the T-cell receptor zeta chain transmembrane and signalling domain. The construct also encodes a truncated/inactive form of CD34 (tCD34) which acts as a marker of transduction. Both the tCD34 and 14C2 CAR were separated by the 2A protease signal from Encephalomyocarditis virus (EMCV), which cleaves the two proteins.
Testing Specificity of M2 CARs In Vitro
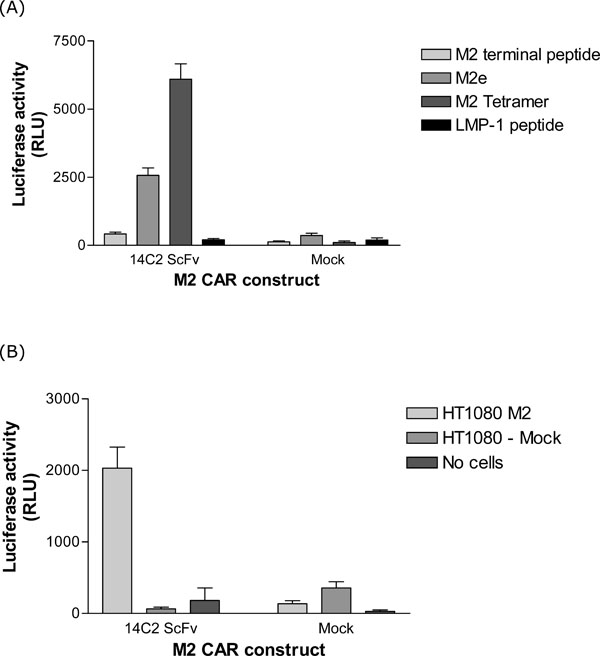
Two in vitro assays were used to determine the specificity of the two M2 CAR constructs. Firstly, an NFAT reporter assay [19Govers C, Sebestyén Z, Berrevoets C, Venselaar H, Debets R. T Cell Receptor Fused to CD3ζ: Transmembrane Domain of CD3ζ Prevents TCR Mis-Pairing, Whereas Complete CD3ζ directs functional TCR expression Open Gene Ther J 2011; 4: 11-22.], which is based on the fact that when T-cells become activated (through recognition of specific antigens), the NFAT transcription factor is upregulated and activated. This can then be measured through its ability to activate a luciferase reporter gene under the control of NFAT control sequences. The M2 CAR construct was transfected into Jurkat T-cells along with a NFAT luciferase reporter plasmid. These Jurkat T-cells were then exposed to M2 specific peptides, or M2 expressing cells (HT1080), and the luciferase activity measured in the supernatant. As shown in Fig. 3A , the 14C2 ScFv CAR resulted in NFAT activation when cells were stimulated with M2e and M2 tetramer peptides, but not with an irrelevant peptide (LMP-1). Significant activation of NFAT in response to M2 expressed on the surface of the HT1080 cell line (Fig. 2) was also observed (Fig. 3B).
, the 14C2 ScFv CAR resulted in NFAT activation when cells were stimulated with M2e and M2 tetramer peptides, but not with an irrelevant peptide (LMP-1). Significant activation of NFAT in response to M2 expressed on the surface of the HT1080 cell line (Fig. 2) was also observed (Fig. 3B).
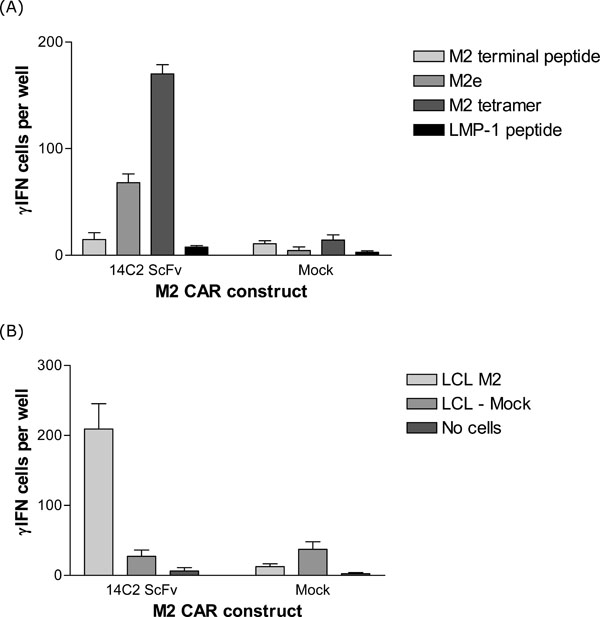
In a second assay, the ELISPOT technique was used to further confirm the specific activation of T-cells with our 14C2 CAR construct in response to stimulation with M2. The ELISPOT assay detects the secretion of γ-Interferon (γ-IFN) from activated T-cells. This assay is quantitative since the γ-IFN produced from each activated T-cell is visualised as a spot on a membrane in a multi-well plate. These spots can be counted and represent the number of activated T-cells in a given population. Human PBMCs were transduced with the retroviral construct encoding the 14C2 CAR. The transduction efficiency as determined by tCD34 expression was 72.5% (data not shown). The transduced PBMCs were then assayed using ELISPOT for γ-IFN production upon stimulation with either M2 peptides or an M2 expressing LCL cell line (LCL114). As shown in Fig. (4 ), significant numbers of transduced T-cells were specifically activated (as measured by γ-IFN release) in response to the specific M2 stimulus.
), significant numbers of transduced T-cells were specifically activated (as measured by γ-IFN release) in response to the specific M2 stimulus.
M2 CAR Mediated Control of Virus Titres in an In Vivo Model
After successfully showing in vitro that the 14C2 CAR was able to recognise and specifically activate transduced T-cells in response to M2 stimulation, 14C2 CAR transduced T-cells were assessed for their ability to modulate virus replication in BALB/c mice infected with the mouse adapted strain of influenza virus A/WSN/33.
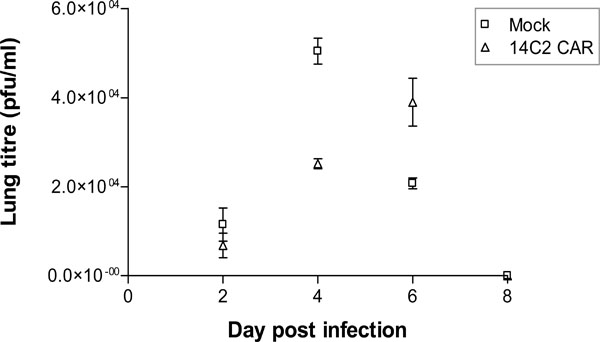
Splenocytes were prepared from BALB/c spleens, activated by stimulation with anti-CD3 antibody, Concanavalin-A and IL-7, and transduced with the retroviral construct encoding 14C2 ScFv CAR. Transduction efficiency was measured though the expression of tCD34 at 88.5% (data not shown). As a control, splenocytes were also transduced with an empty retroviral construct (mock cells). One week post-transduction, the splenocytes (106 cells per mouse) were administered intravenously (IV) via the tail vein. One day following the IV transfer of the cells the mice were infected intranasally with 500pfu of Influenza A/WSN/33. Mice were weighed daily, but showed no significant changes in weight during the course of the experiment (data not shown). On days 2, 4, 6 and 8 pi, groups of 4 mice were sacrificed and lungs removed for titration of virus. The data shown in Fig. (5 ) indicate that in mice receiving the mock transduced splenocytes, A/WSN/33 lung titres increased up to a peak on day 4, before being cleared by day 8. Mice receiving 14C2 M2 CAR transduced splenocytes, however, had a significantly reduced virus lung titre on day 4 (p<0.001), but a higher viral lung titre on day 6 (p<0.001), before being cleared by day 8. H&E staining of lung section revealed significant lymphocyte infiltration and inflammation of the lungs on days 2, 4, and 6 post infection for both 14C2 CAR treated mice and mock treated mice (Fig. 6
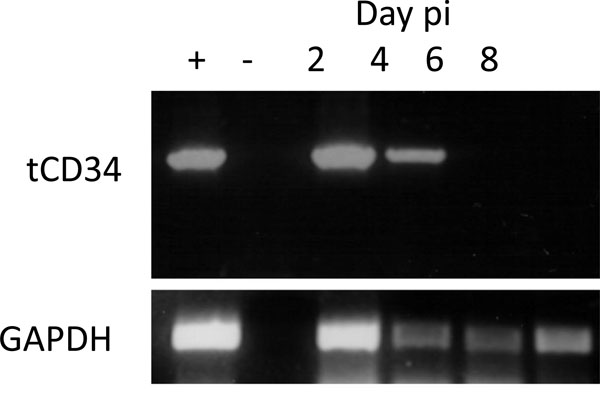
) indicate that in mice receiving the mock transduced splenocytes, A/WSN/33 lung titres increased up to a peak on day 4, before being cleared by day 8. Mice receiving 14C2 M2 CAR transduced splenocytes, however, had a significantly reduced virus lung titre on day 4 (p<0.001), but a higher viral lung titre on day 6 (p<0.001), before being cleared by day 8. H&E staining of lung section revealed significant lymphocyte infiltration and inflammation of the lungs on days 2, 4, and 6 post infection for both 14C2 CAR treated mice and mock treated mice (Fig. 6 ). The presence of tCD34 sequences could be detected in the lungs of 14C2 CAR treated mice by nested PCR on days 2 and 4 post infection (days 3 and 5 post infusion; Fig. 7
). The presence of tCD34 sequences could be detected in the lungs of 14C2 CAR treated mice by nested PCR on days 2 and 4 post infection (days 3 and 5 post infusion; Fig. 7 ), indicating the presence of 14C2 CAR transduced lymphocytes.
), indicating the presence of 14C2 CAR transduced lymphocytes.
DISCUSSION
Influenza A virus poses a continued threat to the world’s population as a result of its pandemic potential and the risk of severe influenza A-induced disease with the emergence of a pandemic strain. Limited range of antiviral drugs and concerns about their effectiveness underscore the importance of developing novel treatment and alternative vaccine strategies. This study aimed to provide evidence that redirecting the T-cell response against the conserved M2e protein of Influenza virus could provide a therapeutic option or contribute to the design of a universal vaccine against multiple Influenza strains.
We generated a Chimeric antigen receptor (CAR) derived from an existing (14C2) hybridoma cell line. The 14C2 monoclonal antibody was produced against the M2e domain from the Influenza A/WSN/33 strain, but is broadly reactive against other Influenza A strains [17Zebedee SL, Lamb RA. Influenza A Virus M2 Protein: Monoclonal Antibody Restriction of Virus Growth and Detection of M2 in Virions J Virol 1988; 62(8): 2762-72.]. When transduced into human T lymphocytes, 14C2 CAR T cells were activated by the M2e target as assessed by NFAT upregulation and interferon-γ production upon encounter in vitro with M2-specific peptides or M2-expressing cell lines (Figs. 3 and 4). In the context of transduced murine splenocytes, the 14C2 CAR delayed peak influenza A titres in lungs of Balb/c mice infected with influenza A/WSN/33 as a small animal model [20Sidwell RW, Smee DF. in vitro and in vivo Assay Systems for Study of Influenza Virus Inhibitors Antiviral Res 2000; 48: 1-16., 21Thomas PG, Keating R, Hulse-Post DJ, et al. Cell-Mediated Protection in Influenza Infection Emerg Infect Dis 2006; 12(1): 48-54.] of virus infection (Fig. 5). The lower titres of virus in the lungs on day 4 correlated to the presence of 14C2 CAR transduced splenocytes (Fig. 7). Although in this model using relatively low titres of Influenza virus, clinical signs of infection (i.e. weight loss) are not observed, the significant reduction in viral lung titres and delay in peak replication may give the immune system additional time to prevent a lethal outcome. A vaccine strategy which could induce both a cell mediated and humoral response to the M2e protein could elicit broad immunity across the different strains of Influenza.
A recent study [22Wise HM, Hutchinson EC, Jagger BW, et al. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain PLoS Pathogens 2012; 8(11): e1002998.] has shown that the expression of an alternative form of M2 by the Influenza A/WSN/33 strain could allow it to escape neutralization by the 14C2 antibody. Specific immune pressure against the M2 protein by the 14C2 CAR splenocytes may therefore reduce viral titres at early time points, but increased expression of alternative forms of M2 (not recognized by 14C2) could allow the virus to escape immune destruction at later times post infection.
The M2e domain has been extensively investigated as a suitable target for a vaccine that could be universally applicable to all influenza A virus strains (including H5N1) because of its highly conserved sequence [23Fiers W, De Filette M, El Bakkouri K, et al. M2e-based universal influenza A vaccine Vaccine 2009; 27(45): 6280-3.-25Ozawa T, Jin A, Tajiri K, et al. Characterization of a fully human monoclonal antibody against extracellular domain of matrix protein 2 of influenza A virus Antiviral Res 2011; 91(3): 283-7.]. Already, clinical studies using candidate M2e-based vaccines have demonstrated immunogenicity and safety in humans [26Turley CB, Rupp RE, Johnson C, et al. Safety and immunogenicity of a recombinant M2e-flagellin influenza vaccine (STF2.4xM2e) in healthy adults Vaccine 2011; 9(32): 5145-2., 27Talbot HK, Rock MT, Johnson C, et al. Immunopotentiation of trivalent influenza vaccine when given with VAX102, a recombinant influenza M2e vaccine fused to the TLR5 ligand flagellin PLoS One 2010; 5(12): e14442.] although some concerns remain as the neutralizing ability of M2e-specific antibodies in humans despite encouraging cross-protective effects observed in animal models [28Wolf AI, Mozdzanowska K, Williams KL, et al. Vaccination with M2e-based multiple antigenic peptides: characterization of the B cell response and protection efficacy in inbred and outbred mice PLoS One 2011; 6(12): e28445.]. In contrast, the use of CAR T cells has focused on their use in adoptive immunotherapy against tumor targets including virus-associated cancers [29Ngo MC, Rooney CM, Howard JM, Heslop HE. Ex vivo gene transfer for improved adoptive immunotherapy of cancer Hum Mol Gene 2011; 20: R93-9., 30Eshhar Z. Adoptive cancer immunotherapy using genetically engineered designer T-cells: First steps into the clinic Curr Opin Mol Ther 2010; 12(1): 55-63.] rather than treatment for acute viral infections such as influenza A. However, our approach provides evidence that redirecting T-cells to Influenza infected cells limits the severity of the infection.
Our results are not only applicable to all influenza A virus strains but also to other virus infections. Future studies are required to characterize the in vivo effect observed in our study and assess the interactions between M2e-specific CARs and influenza A-infected cells in the lung with a view to refine the approach to therapy. Key to such studies is delineation of the immune response to influenza A infection and the manner in which such infection may derail host defenses resulting in the ‘cytokine storm’ phenomenon. It is interesting to note that SCID mice are actually less susceptible to infection with Influenza virus, emphasizing the role of an intact immune system in the pathogenesis of infection [31Palladino G, Mozdzanowska K, Washko G, Gerhard W. Virus-neutralizing antibodies of immunoglobulin G (IgG) but not of IgM or IgA isotypes can cure influenza virus pneumonia in SCID mice J Virol 1995; 69: 2075-81.].
Taken together, we have provided data showing that M2e-specific CAR T-cells can be generated and showed their effectiveness in in vitro and in vivo models. The results support further development M2e as a target for cellular and humoral immunotherapy as a universal treatment for influenza A virus infection and disease.
ACKNOWLEDGEMENTS
The project was funded by CZB/4/521 from the Chief Scientist Office (CSO), Scotland. We are grateful to Prof Reno Debets and Eric Schooten (University of Rotterdam) for retroviral packaging cells and plasmids and advice on transduction of PBMCs and splenocytes.
CONFLICT OF INTERESTS
The authors confirm that this article content has no conflict of interest.
REFERENCES
| [1] | Lamb RA, Krug RM. Orthomyxoviridae: The viruses and their replication In: Knipe DM, Howley PM, Eds. Fields virology. 4th edition. Philadelphia: Lippincott, Williams & Wilkins Publishers 2001; pp. 1487-531. |
| [2] | Wong SSY, Yuen K-Y. Avian Influenza Virus Infections in Humans Chest 2006; 129: 156-68. |
| [3] | von Itzstein M, Wu W-Y, Kok GB, et al. Rational Design of Potent Sialidase-Based Inhibitors of Influenza Virus Replication Nature 1993; 363: 418-23. |
| [4] | Moscona A. Neuraminidase Inhibitors for Influenza N Engl J Med 2005; 353: 1363-73. |
| [5] | Pinto LH, Lamb RA. The M2 Proton Channels of Influenza A and B Viruses J Biol Chem 2006; 281(14): 8997-9000. |
| [6] | Beigel JH, Farrar J, Han AM, et al. Avian Influenza A (H5N1) Infection in Humans N Engl J Med 2005; 353: 1374-85. |
| [7] | Lewis DB. Avian Flu to Human Influenza Ann Rev Med 2006; 57: 139-54. |
| [8] | Gerhard W, Mozdzanowska K, Zharikova D. Prospects for Universal Influenza Virus Vaccine Emerg Infect Dis 2006; 12(4): 569-74. |
| [9] | Doherty PC, Turner SJ, Webby RG, Thomas PG. Influenza and the Challenge for Immunology Nature Immun 2006; 7(5): 449-55. |
| [10] | World Health Organization. ‘Cumulative Number of Confirmed Human Cases of Avian Influenza A/(H5N1) Reported to WHO’ http://www.who.int/influenza/human_animal_interface/EN_GIP_20120706CumulativeNumberH5N1cases.pdf [ACCESSED 12 July 2012]; |
| [11] | de Jong MD, Tran TT, Truong HK, et al. Oseltamivir Resistance During Treatment of Influenza A (H5N1) Infection N Engl J Med 2005; 353: 2667-72. |
| [12] | Mai Le Q, Kiso M, Someya K, et al. Avian flu. isolation of drug-resistant H5N1 virus (brief communication) Nature 2005; 437: 1108. |
| [13] | McCullers JA. The Clinical Need for New Antiviral Drugs Directed against Influenza Virus J Infect Dis 2006; 193: 751-3. |
| [14] | Schumacher TNM. T-Cell-Receptor Gene Therapy Nat Rev Immun 2002; 2: 512-9. |
| [15] | Orentas RJ, Roskopf SJ, Nolan GP, et al. Retroviral Transduction of a T-Cell Receptor Specific for an Epstein-Barr-Virus-Encoded Peptide Clin Immunol 2001; 96: 220-8. |
| [16] | Haynes NM, Trapani JA, Teng MWL, et al. Single-Chain Antigen Recognition Receptors that Costimulate Potent Rejection of Established Experimental Tumours Blood 2002; 100: 3155-63. |
| [17] | Zebedee SL, Lamb RA. Influenza A Virus M2 Protein: Monoclonal Antibody Restriction of Virus Growth and Detection of M2 in Virions J Virol 1988; 62(8): 2762-72. |
| [18] | Cheadle EJ, Hawkins RE, Batha H, O'Neill AL, Dovedi SJ, Gilham DE. Natural Expression of the CD19 Antigen Impacts the Long-Term Engraftment but Not Antitumor Activity of CD19-Specific Engineered T Cells J Immunol 2010; 184: 1885-96. |
| [19] | Govers C, Sebestyén Z, Berrevoets C, Venselaar H, Debets R. T Cell Receptor Fused to CD3ζ: Transmembrane Domain of CD3ζ Prevents TCR Mis-Pairing, Whereas Complete CD3ζ directs functional TCR expression Open Gene Ther J 2011; 4: 11-22. |
| [20] | Sidwell RW, Smee DF. in vitro and in vivo Assay Systems for Study of Influenza Virus Inhibitors Antiviral Res 2000; 48: 1-16. |
| [21] | Thomas PG, Keating R, Hulse-Post DJ, et al. Cell-Mediated Protection in Influenza Infection Emerg Infect Dis 2006; 12(1): 48-54. |
| [22] | Wise HM, Hutchinson EC, Jagger BW, et al. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain PLoS Pathogens 2012; 8(11): e1002998. |
| [23] | Fiers W, De Filette M, El Bakkouri K, et al. M2e-based universal influenza A vaccine Vaccine 2009; 27(45): 6280-3. |
| [24] | Schotsaert M, De Filette M, Fiers W, Saelens X. Universal M2 ectodomain-based influenza A vaccines: preclinical and clinical developments Exp Rev Vaccin 2009; 8(4): 499-508. |
| [25] | Ozawa T, Jin A, Tajiri K, et al. Characterization of a fully human monoclonal antibody against extracellular domain of matrix protein 2 of influenza A virus Antiviral Res 2011; 91(3): 283-7. |
| [26] | Turley CB, Rupp RE, Johnson C, et al. Safety and immunogenicity of a recombinant M2e-flagellin influenza vaccine (STF2.4xM2e) in healthy adults Vaccine 2011; 9(32): 5145-2. |
| [27] | Talbot HK, Rock MT, Johnson C, et al. Immunopotentiation of trivalent influenza vaccine when given with VAX102, a recombinant influenza M2e vaccine fused to the TLR5 ligand flagellin PLoS One 2010; 5(12): e14442. |
| [28] | Wolf AI, Mozdzanowska K, Williams KL, et al. Vaccination with M2e-based multiple antigenic peptides: characterization of the B cell response and protection efficacy in inbred and outbred mice PLoS One 2011; 6(12): e28445. |
| [29] | Ngo MC, Rooney CM, Howard JM, Heslop HE. Ex vivo gene transfer for improved adoptive immunotherapy of cancer Hum Mol Gene 2011; 20: R93-9. |
| [30] | Eshhar Z. Adoptive cancer immunotherapy using genetically engineered designer T-cells: First steps into the clinic Curr Opin Mol Ther 2010; 12(1): 55-63. |
| [31] | Palladino G, Mozdzanowska K, Washko G, Gerhard W. Virus-neutralizing antibodies of immunoglobulin G (IgG) but not of IgM or IgA isotypes can cure influenza virus pneumonia in SCID mice J Virol 1995; 69: 2075-81. |