- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Targeting Wolman Disease and Cholesteryl Ester Storage Disease: Disease Pathogenesis and Therapeutic Development
Francis Aguisanda, Natasha Thorne*, Wei Zheng*
Abstract
Wolman disease (WD) and cholesteryl ester storage disease (CESD) are lysosomal storage diseases (LSDs) caused by a deficiency in lysosomal acid lipase (LAL) due to mutations in the LIPA gene. This enzyme is critical to the proper degradation of cholesterol in the lysosome. LAL function is completely lost in WD while some residual activity remains in CESD. Both are rare diseases with an incidence rate of less than 1/100,000 births for WD and approximate 2.5/100,000 births for CESD. Clinical manifestation of WD includes hepatosplenomegaly, calcified adrenal glands, severe malabsorption and a failure to thrive. As in CESD, histological analysis of WD tissues reveals the accumulation of triglycerides (TGs) and esterified cholesterol (EC) in cellular lysosomes. However, the clinical presentation of CESD is less severe and more variable than WD. This review is to provide an overview of the disease pathophysiology and the current state of therapeutic development for both of WD and CESD. The review will also discuss the application of patient derived iPSCs for further drug discovery.
Article Information

Identifiers and Pagination:
Year: 2017Volume: 11
First Page: 1
Last Page: 18
Publisher Id: CCGTM-11-1
DOI: 10.2174/2213988501711010001
Article History:
Received Date: 30/03/2016Revision Received Date: 20/09/2016
Acceptance Date: 23/11/2016
Electronic publication date: 31/01/2017
Collection year: 2017
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution-Non-Commercial 4.0 International Public License (CC BY-NC 4.0) (https://creativecommons.org/licenses/by-nc/4.0/legalcode), which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to these authors at the National Center for Advancing Translational Sciences, National Institutes of Health, 9800 Medical Center Drive, Bethesda, MD 20892-3375, USA; Tel: (301)217-5251; E-mails: wzheng@mail.nih.gov, natasha.thorne01@gmail.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 30-03-2016 |
Original Manuscript | Targeting Wolman Disease and Cholesteryl Ester Storage Disease: Disease Pathogenesis and Therapeutic Development | |
INTRODUCTION
Lysosomal storage diseases (LSDs) are a group of approximately 50 disorders that result from the defects in lysosomal enzymes or proteins [1Platt F, Boland B, van der Spoel A. Lysosomal storage disorders: the cellular impact of lysosomal function. J Cell Biol 2012; 199: 723-34.
[http://dx.doi.org/10.1083/jcb.201208152] [PMID: 23185029] , 2Xu M, Motabar O, Ferrer M, Marugan JJ, Zheng W, Ottinger EA. Disease models for the development of therapies for lysosomal storage diseases. Ann N Y Acad Sci 2016; 1371(1): 15-29.
[http://dx.doi.org/10.1111/nyas.13052] [PMID: 27144735] ]. Lysosomes play a critical role in maintaining overall cell homeostasis by digesting and recycling lipids and macromolecules through the endosomal-autophagic-lysosomal system. Through macroautophagy, microautophagy, endosome-lysosomal fusion, and chaperone-mediated autophagy, the lysosome is responsible for handling the degradation of dozens of substrates [1Platt F, Boland B, van der Spoel A. Lysosomal storage disorders: the cellular impact of lysosomal function. J Cell Biol 2012; 199: 723-34.
[http://dx.doi.org/10.1083/jcb.201208152] [PMID: 23185029] ]. Functional deficiency of a lysosomal enzyme or protein at any point in a lysosomal degradation pathway can result in accumulation of the lipid or macromolecule upstream of the deficiency, such as unesterified cholesterol in Niemann-Pick disease type C and glycogen in Pompe disease, both of which are LSDs [3Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis 2010; 5: 16.
[http://dx.doi.org/10.1186/1750-1172-5-16] [PMID: 20525256] , 4Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol 2013; 260(4): 951-9.
[http://dx.doi.org/10.1007/s00415-012-6636-x] [PMID: 22926164] ]. Thus, LSDs as a group of diseases can lead to a wide array of clinical presentations and underlying cellular phenotypes depending on the nature of the macromolecule accumulated. Collectively, the approximately 50 LSDs (as a group) have a prevalence of 1 in every 5,000-10,000 live births [5Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999; 281(3): 249-54.
[http://dx.doi.org/10.1001/jama.281.3.249] [PMID: 9918480] ]. Enzyme replacement therapy (ERT), using recombinant wild-type enzyme to replace the mutant enzyme, has been approved for type 1 Gaucher disease [6Brady RO. Benefits from unearthing a biochemical Rosetta Stone. J Biol Chem 2010; 285(53): 41216-21.
[http://dx.doi.org/10.1074/jbc.X110.197954] [PMID: 21030589] ] as well as Fabry disease, MPS1, MPS II (Hunter syndrome), MPS IVA (Morquio A syndrome), MPS VI, Pompe disease, and recently LAL deficiency [2Xu M, Motabar O, Ferrer M, Marugan JJ, Zheng W, Ottinger EA. Disease models for the development of therapies for lysosomal storage diseases. Ann N Y Acad Sci 2016; 1371(1): 15-29.
[http://dx.doi.org/10.1111/nyas.13052] [PMID: 27144735] ]. Substrate reduction therapy (SRT) is another therapeutic approach that reduces the production of the accumulated macromolecule in a given disease. Miglustat, an SRT, was approved in 2006 for treating type 1 Gaucher disease and was later approved for Niemann-Pick type C in Europe [7Pastores GM. Miglustat: substrate reduction therapy for lysosomal storage disorders associated with primary central nervous system involvement. Recent Patents CNS Drug Discov 2006; 1(1): 77-82.
[http://dx.doi.org/10.2174/157488906775245282] [PMID: 18221193] ].
Wolman Disease and Cholesteryl Ester Storage Disease
In 1956, Moshe Wolman and colleagues first reported a case of xanthomatosis with massive calcification of the adrenal glands in a two- year-old infant [8Abramov A, Schorr S, Wolman M. Generalized xanthomatosis with calcified adrenals. AMA J Dis Child 1956; 91(3): 282-6.
[PMID: 13301142] ]. This first patient was admitted to the hospital due to vomiting, a distended abdomen, and a pale yellowish complexion. Upon autopsy of the patient, Wolman also noted the accumulation of foamy lipid droplets in various tissues, including the liver, spleen, intestine, and lymph nodes. The observation of adrenal calcification distinguished this disease from diseases with very similar symptoms, such as Niemann-Pick Type C. In addition to adrenal calcification, hepatosplenomegaly, failure to thrive, and malabsorption are all hallmarks of WD. It is now known that patients with WD have a complete absence of functional Lysosomal Acid Lipase (LAL), caused by mutations in the LIPA gene, which leads to the accumulation of TGs and ECs in the lysosomes in cells of many tissues [9Young EP, Patrick AD. Deficiency of acid esterase activity in Wolmans disease. Arch Dis Child 1970; 45(243): 664-8.
[http://dx.doi.org/10.1136/adc.45.243.664] [PMID: 5477680] , 10Maehira F, Nakada F, Hokama T. Characteristics of acid esterase in Wolmans disease. Biochem Med 1984; 32(3): 322-30.
[http://dx.doi.org/10.1016/0006-2944(84)90037-1] [PMID: 6517876] ]. LAL is an enzyme necessary for the proper intracellular degradation of TGs and CEs, which are delivered to the lysosome via receptor-mediated endocytosis. This complete loss of LAL function is what distinguishes WD from CESD, a similar disease in which patients retain some residual functional LAL [11Porto AF. Lysosomal acid lipase deficiency: diagnosis and treatment of Wolman and Cholesteryl Ester Storage Diseases. Pediatr Endocrinol Rev 2014; 12(Suppl. 1): 125-32.
[PMID: 25345094] , 12Wolman M. Wolman disease and its treatment. Clin Pediatr (Phila) 1995; 34(4): 207-12.
[http://dx.doi.org/10.1177/000992289503400406] [PMID: 7789014] ]. Patients with CESD have widely variable functional levels of LAL (1-12%), resulting in a wide range of clinical phenotypes. CESD may present itself into adulthood, with some patients with less severe forms of the disease likely remaining undiagnosed in their entire life. In contrast to CESD, WD is an infantile onset disease and clinical presentation begins within the first few weeks of life. Markedly more severe than CESD, the progressive course of WD leads to death within the first year.
Epidemiology
WD is rare and is estimated to occur at a rate of less than 1/100,000 newborns [13Wolman Disease. Genetics Home Reference Available at: https://ghrnlmnihgov/condition/wolman-disease 2007.]. WD occurs more frequently in Iranian-Jewish populations (1 in 4,200 newborns) and in areas of the Galilee [14Valles-Ayoub Y, Esfandiarifard S, No D, et al. Wolman disease (LIPA p.G87V) genotype frequency in people of Iranian-Jewish ancestry. Genet Test Mol Biomarkers 2011; 15(6): 395-8.
[http://dx.doi.org/10.1089/gtmb.2010.0203] [PMID: 21291321] ]. Wolman himself pointed out that even in areas with higher than average rates of WD, clinical awareness for WD may not be high, leading to underreporting [12Wolman M. Wolman disease and its treatment. Clin Pediatr (Phila) 1995; 34(4): 207-12.
[http://dx.doi.org/10.1177/000992289503400406] [PMID: 7789014] ]. However, because the clinical phenotype of Wolman Disease is quite severe, it can be readily diagnosed. In contrast, CESD, which can present a range of clinical phenotypes, is more difficult to diagnose and may be significantly under-diagnosed. Although there are few reported cases of CESD, it is estimated that in some European populations as many as 2.5/100,000 people may have biallelic LIPA mutations that result in CESD [15Muntoni S, Wiebusch H, Jansen-Rust M, et al. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol 2007; 27(8): 1866-8.
[http://dx.doi.org/10.1161/ATVBAHA.107.146639] [PMID: 17634524] , 16Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013; 58(6): 1230-43.
[http://dx.doi.org/10.1016/j.jhep.2013.02.014] [PMID: 23485521] ].
Clinical Presentations and Diagnosis
LAL deficiency should be suspected in patients with elevated serum transaminases (AST, ALT, or both), liver fibrosis, cirrhosis, and hepatomegaly. Elevated LDL-cholesterol, triglycerides, and serum total cholesterol are also present in patients [16Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013; 58(6): 1230-43.
[http://dx.doi.org/10.1016/j.jhep.2013.02.014] [PMID: 23485521] , 17Burton BK, Deegan PB, Enns GM, et al. Clinical features of lysosomal acid lipase deficiency. J Pediatr Gastroenterol Nutr 2015; 61(6): 619-25.
[http://dx.doi.org/10.1097/MPG.0000000000000935] [PMID: 26252914] ]. As mentioned previously, WD may be differentiated from CESD based on their differences in disease progress as well as the functional level of LAL in peripheral tissues. Prenatal diagnosis of WD is possible in cases where there is suspected risk due to a family history of WD [18Coates PM, Cortner JA, Mennuti MT, Wheeler JE. Prenatal diagnosis of Wolman disease. Am J Med Genet 1978; 2(4): 397-407.
[http://dx.doi.org/10.1002/ajmg.1320020409] [PMID: 122435] , 19Patrick AD, Willcox P, Stephens R, Kenyon VG. Prenatal diagnosis of Wolmans disease. J Med Genet 1976; 13(1): 49-51.
[http://dx.doi.org/10.1136/jmg.13.1.49] [PMID: 1271426] ]. Though WD symptoms normally take a few weeks to manifest, some abdominal distention in the affected infant may be evident immediately after birth [20Shome DK, Al-Jishi E, Greally JF, Malik N, Zainaldeen HA, Das NS. The Middle-East connection of Wolman Disease. Saudi Med J 2002; 23(5): 597-601.
[PMID: 12070591] ]. Infants are usually admitted into the clinic due to vomiting and diarrhea, a distended abdomen, and an overall failure to thrive [21Crocker AC, Vawter GF, Neuhauser EB, Rosowsky A. Wolmans disease: three new patients with a recently described lipidosis. Pediatrics 1965; 35: 627-40.
[PMID: 14269714] ]. Patients with WD display reduced HDL levels, and increased levels of total cholesterol, triglycerides, and alanine/aspartate transaminase [20Shome DK, Al-Jishi E, Greally JF, Malik N, Zainaldeen HA, Das NS. The Middle-East connection of Wolman Disease. Saudi Med J 2002; 23(5): 597-601.
[PMID: 12070591] , 22Low G, Irwin G, MacPhee G, Robinson P. Characteristic imaging findings in wolman's disease. Clin Radiol Extra 2004; 59: 106-8.
[http://dx.doi.org/10.1016/j.cradex.2004.05.002] -24Shenoy P, Karegowda L, Sripathi S, Mohammed N. Wolman disease in an infant. In: BMJ Case Reports. 2014; 2014. pii: bcr2014203656.]. Moderate jaundice and a fever may be present. A key feature of WD is the enlargement and calcification of the adrenal glands, which can be confirmed via abdominal radiographs or histological examination [25Queloz JM, Capitanio MA, Kirkpatrick JA. Wolmans disease. Roentgen observations in 3 siblings. Radiology 1972; 104(2): 357-9.
[http://dx.doi.org/10.1148/104.2.357] [PMID: 5044990] ]. Although adrenal calcification is a ubiquitous pathological feature of WD, some documented cases of WD patients do not have this affliction [26Schaub J, Janka GE, Christomanou H, et al. Wolmans disease: clinical, biochemical and ultrastructural studies in an unusual case without striking adrenal calcification. Eur J Pediatr 1980; 135(1): 45-53.
[http://dx.doi.org/10.1007/BF00445892] [PMID: 7449788] ]. CT scans of the abdomen can reveal distended bowel loops [22Low G, Irwin G, MacPhee G, Robinson P. Characteristic imaging findings in wolman's disease. Clin Radiol Extra 2004; 59: 106-8.
[http://dx.doi.org/10.1016/j.cradex.2004.05.002] ] and biopsy of the small intestine can show infiltration of the lamina propria by foamy macrophages filled with cholesteryl esters and triglycerides. The liver, spleen, lymph nodes, and tonsils may also be enlarged due to accumulation of triglycerides and cholesteryl esters in lysosomes of infiltrating macrophages and other cells [27Hill SC, Hoeg JM, Dwyer AJ, Vucich JJ, Doppman JL. CT findings in acid lipase deficiency: wolman disease and cholesteryl ester storage disease. J Comput Assist Tomogr 1983; 7(5): 815-8.
[http://dx.doi.org/10.1097/00004728-198310000-00011] [PMID: 6886132] , 28Du H, Schiavi S, Levine M, Mishra J, Heur M, Grabowski GA. Enzyme therapy for lysosomal acid lipase deficiency in the mouse. Hum Mol Genet 2001; 10(16): 1639-48.
[http://dx.doi.org/10.1093/hmg/10.16.1639] [PMID: 11487567] ]. These macrophages also display cholesterol-like crystals [29Lake BD, Patrick AD. Wolmans disease: deficiency of E600-resistant acid esterase activity with storage of lipids in lysosomes. J Pediatr 1970; 76(2): 262-6.
[http://dx.doi.org/10.1016/S0022-3476(70)80172-X] [PMID: 5410174] ]. WD-affected livers appear yellow and greasy and a more detailed histological examination can reveal fibrosis and large lipid-filled Kupffer cells [30Boldrini R, Devito R, Biselli R, Filocamo M, Bosman C. Wolman disease and cholesteryl ester storage disease diagnosed by histological and ultrastructural examination of intestinal and liver biopsy. Pathol Res Pract 2004; 200(3): 231-40.
[http://dx.doi.org/10.1016/j.prp.2003.11.001] [PMID: 15200275] ]. The spleen can be over 20 times the normal size, and filled with numerous lipid-filled vacuoles [31Wolman M, Sterk VV, Gatt S, Frenkel M. Primary familial xanthomatosis with involvement and calcification of the adrenals. Report of two more cases in siblings of a previously described infant. Pediatrics 1961; 28: 742-57.
[PMID: 14008104] ]. Many of these symptoms can also be associated with other lysosomal storage diseases such as Niemann-Pick Type C and Gaucher disease, but a confident diagnosis may be made by assaying patient tissue samples for LAL activity and by examining the patient’s genotype. Biochemical assays for measurements of LAL activity can also be used to differentiate WD from CESD (Table 1). In WD, secondary abnormalities due to malabsorption may occur, such as anemia and abnormal liver function [32Röyttä M, Fagerlund AS, Toikkanen S, et al. Wolman disease: morphological, clinical and genetic studies on the first Scandinavian cases. Clin Genet 1992; 42(1): 1-7.
[http://dx.doi.org/10.1111/j.1399-0004.1992.tb03125.x] [PMID: 1516222] ]. A paucity of fat in subcutaneous tissue may be noted, and generalized osteoporosis, though rare, is possible [25Queloz JM, Capitanio MA, Kirkpatrick JA. Wolmans disease. Roentgen observations in 3 siblings. Radiology 1972; 104(2): 357-9.
[http://dx.doi.org/10.1148/104.2.357] [PMID: 5044990] ]. The malabsorption, however, is usually so severe that normal enteral nutrition may not be enough to provide adequate nutrients. Hyperalimentation via intravenous drip has been found to be only mildly effective [33Kikuchi M, Igarashi K, Noro T, Igarashi Y, Hirooka M, Tada K. Evaluation of jejunal function in Wolmans disease. J Pediatr Gastroenterol Nutr 1991; 12(1): 65-9.
[http://dx.doi.org/10.1097/00005176-199101000-00014] [PMID: 2061782] ]. Patients with WD do not live beyond the first year of life [34Grabowski G, Du H, Charnas L. Lysosomal acid lipase deficiencies: the wolman disease/cholesteryl ester storage disease spectrum the online metabolic & molecular bases of inherited disease. New York: McGraw-Hill 2013; p. 142.].

The clinical presentations of CESD are highly variable due to the range of residual LAL activity levels that cause the disease. Clinical presentation may occur as early as infancy or as late as the fifth and sixth decades of life; sometimes CESD is diagnosed completely by accident [35Chatrath H, Keilin S, Attar BM. Cholesterol ester storage disease (CESD) diagnosed in an asymptomatic adult. Dig Dis Sci 2009; 54(1): 168-73.
[http://dx.doi.org/10.1007/s10620-008-0310-2] [PMID: 18478331] ]. Patients with severe CESD may present with Wolman Disease-like symptoms, such as diarrhea and failure to thrive [16Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013; 58(6): 1230-43.
[http://dx.doi.org/10.1016/j.jhep.2013.02.014] [PMID: 23485521] ]. Patients typically have hepatomegaly and liver dysfunction with liver biopsies revealing microvesicular steatosis in the hepatocytes [36Hůlková H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology 2012; 60(7): 1107-13.
[http://dx.doi.org/10.1111/j.1365-2559.2011.04164.x] [PMID: 22621222] ]. This liver dysfunction is associated with the elevation of serum transaminases, such as alanine aminotransferase (ALT) and aspartate aminotransferase (AST). Patients with CESD have increased levels of triglycerides and serum total/LDL cholesterol, the latter of which can cause accelerated atherosclerosis [16Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013; 58(6): 1230-43.
[http://dx.doi.org/10.1016/j.jhep.2013.02.014] [PMID: 23485521] , 37Kwiterovich PO Jr. The metabolic pathways of high-density lipoprotein, low-density lipoprotein, and triglycerides: a current review. Am J Cardiol 2000; 86(12A): 5L-10L.
[http://dx.doi.org/10.1016/S0002-9149(00)01461-2] [PMID: 11374859] ]. Patients with CESD have reduced serum HDL levels. Increased infiltration of the sinusoids by Kupffer cells and macrophages also occurs, both of which show cytoplasmic storage of foamy, fatty material [38Drebber U, Andersen M, Kasper HU, Lohse P, Stolte M, Dienes HP. Severe chronic diarrhea and weight loss in cholesteryl ester storage disease: a case report. World J Gastroenterol 2005; 11(15): 2364-6.
[http://dx.doi.org/10.3748/wjg.v11.i15.2364] [PMID: 15818756] ]. Lipid deposition in the liver can lead to cirrhosis [30Boldrini R, Devito R, Biselli R, Filocamo M, Bosman C. Wolman disease and cholesteryl ester storage disease diagnosed by histological and ultrastructural examination of intestinal and liver biopsy. Pathol Res Pract 2004; 200(3): 231-40.
[http://dx.doi.org/10.1016/j.prp.2003.11.001] [PMID: 15200275] ]. Compared with WD, the cholesteryl ester content of the liver tissue is much higher in CESD. In general, the accumulation of cholesteryl esters in tissues is higher than that of triglycerides due a lower cholesteryl ester affinity for the remaining functional LAL [16Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013; 58(6): 1230-43.
[http://dx.doi.org/10.1016/j.jhep.2013.02.014] [PMID: 23485521] , 39Groener JE, Bax W, Stuani C, Pagani F. Difference in substrate specificity between human and mouse lysosomal acid lipase: low affinity for cholesteryl ester in mouse lysosomal acid lipase. Biochim Biophys Acta 2000; 1487(2-3): 155-62.
[http://dx.doi.org/10.1016/S1388-1981(00)00091-3] [PMID: 11018468] ]. Needle shaped autofluorescent crystals can be seen in CESD hepatocytes. Though rare, some patients have splenomegaly and adrenal calcification [40Beaudet AL, Ferry GD, Nichols BL Jr, Rosenberg HS. Cholesterol ester storage disease: clinical, biochemical, and pathological studies. J Pediatr 1977; 90(6): 910-4.
[http://dx.doi.org/10.1016/S0022-3476(77)80557-X] [PMID: 859064] , 41Edelstein RA, Filling-Katz MR, Pentchev P, et al. Cholesteryl ester storage disease: a patient with massive splenomegaly and splenic abscess. Am J Gastroenterol 1988; 83(6): 687-92.
[PMID: 3376925] ]. CESD-related mortality is often due to liver failure and/or secondary accelerated atherosclerotic disease [16Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013; 58(6): 1230-43.
[http://dx.doi.org/10.1016/j.jhep.2013.02.014] [PMID: 23485521] ].
When diagnosing CESD, it is critical to differentiate between patients with true CESD and those with non-alcoholic fatty liver disease (NAFLD). Patients with NAFLD may also present with elevated aminotransferase levels as well as hepatic steatosis. To confirm a CESD diagnosis, Hulkova et al. recommend a more sophisticated histological evaluation of a liver biopsy. The use of fixed, paraffin-embedded and unfixed frozen liver samples can enable the recognition of accumulated lipids that are localized to the lysosome. One may also use additional immunohistochemical analyses with lysosomal markers to confirm lysosomal accumulation (LAMP1, LAMP2, etc.) [36Hůlková H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology 2012; 60(7): 1107-13.
[http://dx.doi.org/10.1111/j.1365-2559.2011.04164.x] [PMID: 22621222] ]. Unlike CESD, NAFLD liver cells display macrosteatosis as well as masses of lipid occupying the whole cell cytoplasm.
A diagnosis of WD and CESD requires a biochemical analysis of functional LAL levels in patient cells. Levels of LAL activity can be detected in easily obtainable patient tissue samples (i.e. skin fibroblasts or blood leukocytes) via a fluorometric enzyme activity assay using 4-methylumbelliferone (4-MU) palmitate as a substrate [42Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta 2012; 413(15-16): 1207-10.
[http://dx.doi.org/10.1016/j.cca.2012.03.019] [PMID: 22483793] , 43Dairaku T, Iwamoto T, Nishimura M, Endo M, Ohashi T, Eto Y. A practical fluorometric assay method to measure lysosomal acid lipase activity in dried blood spots for the screening of cholesteryl ester storage disease and Wolman disease. Mol Genet Metab 2014; 111(2): 193-6.
[http://dx.doi.org/10.1016/j.ymgme.2013.11.003] [PMID: 24295952] ]. When 4-MU palmitate is cleaved, a fluorescent product is produced that can be measured via a fluorescence reader. Because 4-MU palmitate cleavage is not specific to LAL, a specific inhibitor is used to differentiate between general lipase activity and LAL activity. This assay can also be performed in amniotic fluid cells for prenatal screening [44Christomanou H, Cáp C. Prenatal monitoring for Wolmans disease in a pregnancy at risk: first case in the Federal Republic of Germany. Hum Genet 1981; 57(4): 440-1.
[http://dx.doi.org/10.1007/BF00281702] [PMID: 7286988] , 45Desai PK, Astrin KH, Thung SN, et al. Cholesteryl ester storage disease: pathologic changes in an affected fetus. Am J Med Genet 1987; 26(3): 689-98.
[http://dx.doi.org/10.1002/ajmg.1320260324] [PMID: 3565483] ]. While assaying for LAL activity may give a clear diagnosis of LAL deficiency, a definitive diagnosis may be made by the exonic sequencing of the LIPA gene. Additionally, exonic sequencing may be required when measuring LAL activity alone gives an ambiguous diagnosis on the LAL deficiency spectrum [46Anderson RA, Bryson GM, Parks JS. Lysosomal acid lipase mutations that determine phenotype in Wolman and cholesterol ester storage disease. Mol Genet Metab 1999; 68(3): 333-45.
[http://dx.doi.org/10.1006/mgme.1999.2904] [PMID: 10562460] ]. Anderson et al. showed that in contrast to CESD, WD patients have no functioning levels of LAL and their mutations predict a loss of intact LAL protein [46Anderson RA, Bryson GM, Parks JS. Lysosomal acid lipase mutations that determine phenotype in Wolman and cholesterol ester storage disease. Mol Genet Metab 1999; 68(3): 333-45.
[http://dx.doi.org/10.1006/mgme.1999.2904] [PMID: 10562460] ]. Each of the five WD mutations identified in their study were nonsense, resulting in the cessation of translation of LAL. Subsequently, no intact LAL protein was detected in immunoblots from these patients. The sequencing of LIPA in CESD patients revealed the presence of an exon 8 splice junction mutation (c.894G>A; E8SJM), the most common mutation associated with CESD [47Scott SA, Liu B, Nazarenko I, et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology 2013; 58(3): 958-65.
[http://dx.doi.org/10.1002/hep.26327] [PMID: 23424026] ]. The reported mutations of LIPA for both WD and CESD are summarized in Table 2.
Etiology
LAL is responsible for the degradation of EC as well as mono-, di- and TGs in lysosomes. It is present in nearly all cells with the exception of erythrocytes [48Lohse P, Chahrokh-Zadeh S, Lohse P, Seidel D. Human lysosomal acid lipase/cholesteryl ester hydrolase and human gastric lipase: identification of the catalytically active serine, aspartic acid, and histidine residues. J Lipid Res 1997; 38(5): 892-903.
[PMID: 9186907] ]. CEs and TGs are bound to low density lipoproteins (LDL), which are taken into the cell by receptor-mediated endocytosis. These endocytic vesicles then fuse with the lysosome. Once in the lysosome, CEs and TGs are cleaved by LAL and exit into the cytosol via two lysosomal proteins, Niemann-Pick type C1 (NPC1) and NPC type C2 (NPC-2) [49Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J Biol Chem 1975; 250(21): 8487-95.
[PMID: 172501] ]. The resulting unesterified cholesterol can then be used as a substrate for steroidogenesis or reesterified by acyl CoA: cholesterol acyltransferase (ACAT) for other uses [50Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking. J Lipid Res 2011; 52(12): 2111-35.
[http://dx.doi.org/10.1194/jlr.R016675] [PMID: 21976778] ]. In WD patients, CEs and TGs are not cleaved which results in the accumulation of these macromolecules in the lysosome. Intriguingly, experiments in WD fibroblasts have shown that this accumulation is due to the intake of external LDL into the cell whereas endogenously produced CEs and TGs may not be the main sources of lipids for the accumulation [51Salvayre R, Negre A, Maret A, Radom J, Douste-Blazy L. Extracellular origin of the lipid lysosomal storage in cultured fibroblasts from Wolmans disease. Eur J Biochem 1987; 170(1-2): 453-8.
[http://dx.doi.org/10.1111/j.1432-1033.1987.tb13721.x] [PMID: 3691534] ]. In the adrenal glands, a lack of free cholesterol available for steroidogenesis results in adrenal insufficiency. The lack of cholesterol excretion into the cytosol is interpreted as a deficiency in intracellular cholesterol, so the sterol regulatory element-binding protein 1c/2 (SREBP-1c/2) system upregulates genes related to the expression of LDL receptors and synthesis of fatty acids, thus causing additional accumulation [52Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiencyan under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014; 235(1): 21-30.
[http://dx.doi.org/10.1016/j.atherosclerosis.2014.04.003] [PMID: 24792990] ]. Together, ACAT and LAL maintain equilibrium between unesterified and esterified cholesterols in cells.
Mutations in the LIPA gene are located in human chromosome 10q23.2-q23.3 and are autosomal recessive [34Grabowski G, Du H, Charnas L. Lysosomal acid lipase deficiencies: the wolman disease/cholesteryl ester storage disease spectrum the online metabolic & molecular bases of inherited disease. New York: McGraw-Hill 2013; p. 142.]. The LIPA gene contains 10 exons, 9 introns and is 36kb long. As previously mentioned, WD patients possess less than 1% of normal LAL levels and CESD is characterized by 1-12% of normal LAL activity [16Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013; 58(6): 1230-43.
[http://dx.doi.org/10.1016/j.jhep.2013.02.014] [PMID: 23485521] ]. These differences in levels of functional LAL are dependent on the types of mutations at the LIPA locus. To date, over a dozen different mutations in the LIPA gene have been reported for WD. The mutations are diverse and are usually nonsense mutations with deletions or insertions in the LIPA gene that ultimately result in the absence of a functional enzyme [34Grabowski G, Du H, Charnas L. Lysosomal acid lipase deficiencies: the wolman disease/cholesteryl ester storage disease spectrum the online metabolic & molecular bases of inherited disease. New York: McGraw-Hill 2013; p. 142.-53Zschenker O, Jung N, Rethmeier J, et al. Characterization of lysosomal acid lipase mutations in the signal peptide and mature polypeptide region causing Wolman disease. J Lipid Res 2001; 42(7): 1033-40.
[PMID: 11441129] ]. For example, a G to A substitution at nucleotide 347 in exon 4 converts a tryptophan codon into a termination codon in WD patients. CESD is generally caused by missense mutations in the LIPA gene, and are heteroallelic or homoallelic with other mutations in this gene, generating moderately functioning LAL from either or both alleles [34Grabowski G, Du H, Charnas L. Lysosomal acid lipase deficiencies: the wolman disease/cholesteryl ester storage disease spectrum the online metabolic & molecular bases of inherited disease. New York: McGraw-Hill 2013; p. 142., 54Aslanidis C, Ries S, Fehringer P, Büchler C, Klima H, Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics 1996; 33(1): 85-93.
[http://dx.doi.org/10.1006/geno.1996.0162] [PMID: 8617513] ]. A mutation common in CESD is the G -> A mutation at position -1 of the exon 8 splice donor site (c.894G>A; E8SJM) and accounts for an estimated 60% of all CESD cases. This mutation results in a transcript with no LAL activity; however, the defect allows for 3-5% of LAL to be normally spliced [55Pagani F, Pariyarath R, Garcia R, et al. New lysosomal acid lipase gene mutants explain the phenotype of Wolman disease and cholesteryl ester storage disease. J Lipid Res 1998; 39(7): 1382-8.
[PMID: 9684740] , 56Klima H, Ullrich K, Aslanidis C, Fehringer P, Lackner KJ, Schmitz G. A splice junction mutation causes deletion of a 72-base exon from the mRNA for lysosomal acid lipase in a patient with cholesteryl ester storage disease. J Clin Invest 1993; 92(6): 2713-8.
[http://dx.doi.org/10.1172/JCI116888] [PMID: 8254026] ]. Compound heterozygosity can also result in CESD. For example, some CESD patients possess the G to A mutation at the exon 8 splice donor site position -1 on one allele and a nonsense mutation on the other allele [57Ameis D, Brockmann G, Knoblich R, et al. A 5 splice-region mutation and a dinucleotide deletion in the lysosomal acid lipase gene in two patients with cholesteryl ester storage disease. J Lipid Res 1995; 36(2): 241-50.
[PMID: 7751811] ].
While the crystal structure of LAL has yet to be solved, Roussel et al. proposed several theories on how LAL mutations may affect the LAL structure based on their solved crystal structure of human gastric lipase, both of which share 59% identity and 75% sequence homology [58]. A Gly-66->Val mutation would result the enzyme’s valine side chain both blocking the active site serine 153 and the triglyceride binding site. Additionally, a Leu-273->Ser mutation would create a new glycosylation site, which may also prevent substrate binding [59Pagani F, Garcia R, Pariyarath R, et al. Expression of lysosomal acid lipase mutants detected in three patients with cholesteryl ester storage disease. Hum Mol Genet 1996; 5(10): 1611-7.
[http://dx.doi.org/10.1093/hmg/5.10.1611] [PMID: 8894696] ].
Complications of WD that result from the accumulation of CE and TG are related to malabsorption. Under normal circumstances, triglycerides are absorbed by the intestinal lumen and degraded in the intestinal villi. In WD, the accumulation of foamy macrophages and Kupffer cells in the intestinal villi is hypothesized to inhibit proper nutrient absorption due to the clogging of the mucosa by non-degraded triglycerides [12Wolman M. Wolman disease and its treatment. Clin Pediatr (Phila) 1995; 34(4): 207-12.
[http://dx.doi.org/10.1177/000992289503400406] [PMID: 7789014] ]. For example, unabsorbed triolein can be seen in the stool samples of WD patients [60Eto Y, Kitagawa T. Wolmans disease with hypolipoproteinemia and acanthocytosis: clinical and biochemical observations. J Pediatr 1970; 77(5): 862-7.
[http://dx.doi.org/10.1016/S0022-3476(70)80248-7] [PMID: 5504079] ]. The strongest LAL activity in rats was found in the regions of intestinal villi that most involve the absorption and transport of dietary lipids, so LAL mutations would impair the function of these villi [61Rao RH, Mansbach CM II. Acid lipase in rat intestinal mucosa: physiological parameters. Biochim Biophys Acta 1990; 1043(3): 273-80.
[http://dx.doi.org/10.1016/0005-2760(90)90027-U] [PMID: 2322572] ]. In an attempt to circumvent malabsorption, a course of parenteral hyperalimentation show to slow patient deterioration. While this approach is only palliative, it may be useful for extending a patient’s lifespan long enough to consider more long-term treatment options [62Meyers W, Hoeg J, Demosky S, Herbst J, Brewer H. The use of parenteral hyperalimentation and elemental formula feeding in the treatment of wolman disease. Nutr Res 1985; 5: 423-9.
[http://dx.doi.org/10.1016/S0271-5317(85)80226-8] ].
Additionally, high levels of oxidized CEs have been found in WD tissues, suggesting either high levels of innate lipid peroxidation or the increased intake of oxidized LDL [63Assmann G, Fredrickson DS, Sloan HR, Fales HM, Highet RJ. Accumulation of oxygenated steryl esters in Wolmans disease. J Lipid Res 1975; 16(1): 28-38.
[PMID: 162929] ]. It has been hypothesized that lipid peroxidation derivatives are involved in the pathogenesis of WD [64Fitoussi G, Nègre-Salvayre A, Pieraggi MT, Salvayre R. New pathogenetic hypothesis for Wolman disease: possible role of oxidized low-density lipoproteins in adrenal necrosis and calcification. Biochem J 1994; 301(Pt 1): 267-73.
[http://dx.doi.org/10.1042/bj3010267] [PMID: 8037680] ]. In cultured adrenal cells, oxidized LDL caused increased levels of intracellular calcium that were subsequently deposited in damaged/dead cells [65Nègre-Salvayre A, Salvayre R. UV-treated lipoproteins as a model system for the study of the biological effects of lipid peroxides on cultured cells. 4. Calcium is involved in the cytotoxicity of UV-treated LDL on lymphoid cell lines. Biochim Biophys Acta 1992; 1123(2): 207-15.
[http://dx.doi.org/10.1016/0005-2760(92)90113-A] [PMID: 1739749] ]. This finding suggests a plausible mechanism by which adrenal calcification occurs in WD patients. In addition to the adrenal glands, lysosomal accumulation of CEs and TGs occurs in the cells of many other tissues, including the small intestine, spleen, lymph nodes, thymus, alveoli in the lungs, and the liver [66Marshall WC, Ockenden BG, Fosbrooke AS, Cumings JN. Wolmans disease. A rare lipidosis with adrenal calcification. Arch Dis Child 1969; 44(235): 331-41.
[http://dx.doi.org/10.1136/adc.44.235.331] [PMID: 5785183] ]. In several of these organs, tissues swell with “foamy macrophages” that are themselves engorged with TG and CEs [30Boldrini R, Devito R, Biselli R, Filocamo M, Bosman C. Wolman disease and cholesteryl ester storage disease diagnosed by histological and ultrastructural examination of intestinal and liver biopsy. Pathol Res Pract 2004; 200(3): 231-40.
[http://dx.doi.org/10.1016/j.prp.2003.11.001] [PMID: 15200275] , 67Lowden JA, Barson AJ, Wentworth P. Wolmans disease: a microscopic and biochemical study showing accumulation of ceroid and esterified cholesterol. Can Med Assoc J 1970; 102(4): 402-5.
[PMID: 5414926] , 68Lough J, Fawcett J, Wiegensberg B. Wolmans disease. An electron microscopic, histochemical, and biochemical study. Arch Pathol 1970; 89(2): 103-10.
[PMID: 5412920] ]. This macrophage accumulation results in cirrhosis of the liver as well as the impaired ability of the intestinal villi to absorb nutrients. The combination of cachexia, malnutrition and hepatic failure due to cirrhosis of the liver ultimately causes death in WD [11Porto AF. Lysosomal acid lipase deficiency: diagnosis and treatment of Wolman and Cholesteryl Ester Storage Diseases. Pediatr Endocrinol Rev 2014; 12(Suppl. 1): 125-32.
[PMID: 25345094] , 23Sadhukhan M, Saha A, Vara R, Bhaduri B. Infant case of lysosomal acid lipase deficiency: Wolmans disease. BMJ Case Rep 2014; 2014: 1-3.
[http://dx.doi.org/10.1136/bcr-2013-202652] [PMID: 24832708] ].
Accumulation of CEs and TGs has been observed in the central nervous system (CNS) in cells such as astrocytes and oligodendrocytes [69Byrd JC III, Powers JM. Wolmans disease: ultrastructural evidence of lipid accumulation in central and peripheral nervous systems. Acta Neuropathol 1979; 45(1): 37-42.
[http://dx.doi.org/10.1007/BF00691802] [PMID: 216225] ]. Infants born with WD show cognitive decline after initial onset of the disease, but it is not known whether this is due to the accumulation in CNS or is a secondary consequence of the disease due to malabsorption. The accumulation of TGs and CEs can also be found in the neurons of the myenteric plexus, causing severe neuronal damage, and may contribute to the gastrointestinal pathology found in WD patients [70Kamoshita S, Landing BH. Distribution of lesions in myenteric plexus and gastrointestinal mucosa in lipidoses and other neurologic disorders of children. Am J Clin Pathol 1968; 49(3): 312-8.
[http://dx.doi.org/10.1093/ajcp/49.3.312] [PMID: 5645093] ]. Additionally, some investigators have noted impaired myelination of neurons in the brain, a possible consequence of impaired CE and TG metabolism [71Stein J, Garty BZ, Dror Y, Fenig E, Zeigler M, Yaniv I. Successful treatment of Wolman disease by unrelated umbilical cord blood transplantation. Eur J Pediatr 2007; 166(7): 663-6.
[http://dx.doi.org/10.1007/s00431-006-0298-6] [PMID: 17033804] ]. While the perturbation of normal lysosomal pathways often results in neurodegeneration for many LSDs, it is not clear if this is characteristic of WD [69Byrd JC III, Powers JM. Wolmans disease: ultrastructural evidence of lipid accumulation in central and peripheral nervous systems. Acta Neuropathol 1979; 45(1): 37-42.
[http://dx.doi.org/10.1007/BF00691802] [PMID: 216225] , 72Osellame LD, Duchen MR. Quality control gone wrong: mitochondria, lysosomal storage disorders and neurodegeneration. Br J Pharmacol 2014; 171(8): 1958-72.
[http://dx.doi.org/10.1111/bph.12453] [PMID: 24116849] , 73Guazzi GC, Martin JJ, Philippart M, et al. Wolmans disease. Eur Neurol 1968; 1(6): 334-62.
[http://dx.doi.org/10.1159/000113673] [PMID: 5711398] ]. The high infantile mortality associated with WD prevents the assessment of long-term neurological damage.
CESD is a highly variable disease and its clinical course is poorly defined. The literature contains reports on only the most severe of these cases; thus, an underreporting of milder forms of the disease may skew our perception of its typical symptoms [34Grabowski G, Du H, Charnas L. Lysosomal acid lipase deficiencies: the wolman disease/cholesteryl ester storage disease spectrum the online metabolic & molecular bases of inherited disease. New York: McGraw-Hill 2013; p. 142.]. This underreporting also affects how we perceive CESD’s lethality, which is often due to liver failure [74Gasche C, Aslanidis C, Kain R, et al. A novel variant of lysosomal acid lipase in cholesteryl ester storage disease associated with mild phenotype and improvement on lovastatin. J Hepatol 1997; 27(4): 744-50.
[http://dx.doi.org/10.1016/S0168-8278(97)80092-X] [PMID: 9365051] ]. The most common presorted symptoms of the disease are hepatomegaly and hyperlipidemia. As described for WD, hyperlipidemia likely results from the improper processing of triglycerides and cholesteryl esters due to low levels of LAL activity. Hepatomegaly is caused by both the accumulation of lipid vacuoles in liver tissue and by the infiltration of tissues by lipid filled macrophages and Kupffer cells. In a review of 135 cases of CESD reported in the literature, Bernstein et al. noted that all reported cases had significant liver disease that progressed into micronodular cirrhosis and liver failure [16]. Although rare, lipid accumulation may also cause chronic diarrhea and chronic malabsorption [38Drebber U, Andersen M, Kasper HU, Lohse P, Stolte M, Dienes HP. Severe chronic diarrhea and weight loss in cholesteryl ester storage disease: a case report. World J Gastroenterol 2005; 11(15): 2364-6.
[http://dx.doi.org/10.3748/wjg.v11.i15.2364] [PMID: 15818756] ]. In a few cases of CESD, neurological manifestations occurred in a 4-year-old boy and his 22-month old sibling. One had periods of headache, vertigo, and loss of consciousness. Both had external opthalmoplegia and bilateral ptosis [75Bindu PS, Taly AB, Christopher R, et al. Cholesterol ester storage disease with unusual neurological manifestations in two siblings: a report from South India. J Child Neurol 2007; 22(12): 1401-4.
[http://dx.doi.org/10.1177/0883073807307104] [PMID: 18174560] ]. The pathological cause for such manifestations remains unclear.
Therapeutics and New Drug Development
Hematopoietic Stem Cell transplantation, Bone Marrow Transplantation, and Liver Transplantation
A moderately successful treatment for WD is hematopoietic stem cell transplantation (HSCT) [76Tolar J, Petryk A, Khan K, et al. Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease. Bone Marrow Transplant 2009; 43(1): 21-7.
[http://dx.doi.org/10.1038/bmt.2008.273] [PMID: 18776925] , 77Krivit W, Peters C, Dusenbery K, et al. Wolman disease successfully treated by bone marrow transplantation. Bone Marrow Transplant 2000; 26(5): 567-70.
[http://dx.doi.org/10.1038/sj.bmt.1702557] [PMID: 11019848] ]. In a cohort of 4 patients, the donor hematopoietic stem cells were infused into the affected infants. Of the four infants that received this therapy, two lived to the ages of 4 and 11 [76Tolar J, Petryk A, Khan K, et al. Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease. Bone Marrow Transplant 2009; 43(1): 21-7.
[http://dx.doi.org/10.1038/bmt.2008.273] [PMID: 18776925] ]. A reduction in the number of hypertrophic Kupffer cells and the amelioration of hepatosplenomegaly were observed [78Yanir A, Allatif MA, Weintraub M, Stepensky P. Unfavorable outcome of hematopoietic stem cell transplantation in two siblings with Wolman disease due to graft failure and hepatic complications. Mol Genet Metab 2013; 109(2): 224-6.
[http://dx.doi.org/10.1016/j.ymgme.2013.03.007] [PMID: 23583223] ]. Another report showed that some normal LAL enzyme activity was detected in peripheral cells from an infant WD patient who received HSCT, although the patient was lost due to sepsis and multi-organ failure [79Gramatges MM, Dvorak CC, Regula DP, Enns GM, Weinberg K, Agarwal R. Pathological evidence of Wolmans disease following hematopoietic stem cell transplantation despite correction of lysosomal acid lipase activity. Bone Marrow Transplant 2009; 44(7): 449-50.
[http://dx.doi.org/10.1038/bmt.2009.57] [PMID: 19308038] ]. A post-mortem analysis of this patient’s liver showed a lack of donor-derived macrophages; thus, the patient perished from complications due to WD and surgery before donor-derived macrophage engraftment in the liver occurred. Because a full correction of enzyme deficiency in recipient tissues can take as long as a year, pathological tissue damage already inflicted by the disease can prove to be fatal. Because tissue damage in WD patients arise quickly, it will be essential to find therapies that can be rapidly deployed to the affected tissues to avoid the high risk of complication associated with HSCT. Krivit and colleagues reported a successful case of bone marrow transplantation (BMT), in which bone marrow-derived hematopoietic cells are transferred to the patient, resulting in normalization of cholesterol levels [77Krivit W, Peters C, Dusenbery K, et al. Wolman disease successfully treated by bone marrow transplantation. Bone Marrow Transplant 2000; 26(5): 567-70.
[http://dx.doi.org/10.1038/sj.bmt.1702557] [PMID: 11019848] ]. Liver dysfunction leading to fibrosis was also prevented. Despite this success, four other patients in the same cohort died due to either WD or complications arising from the procedure. Data from other diseases suggests that BMT may stop the progression of hepatic fibrosis and reduce it in some cases [80Muretto P, Del Fiasco S, Angelucci E, De Rosa F, Lucarelli G. Bone marrow transplantation in thalassemia: modifications of hepatic iron overload and associated lesions after long-term engrafting. Liver 1994; 14(1): 14-24.
[http://dx.doi.org/10.1111/j.1600-0676.1994.tb00002.x] [PMID: 8177025] ]. BMT may provide the best approach for reducing hepatic fibrosis because it has been reported that WD patients who received a liver transplantation eventually resumed cirrhosis of the liver [81Krivit W, Freese D, Chan KW, Kulkarni R. Wolmans disease: a review of treatment with bone marrow transplantation and considerations for the future. Bone Marrow Transplant 1992; 10(Suppl. 1): 97-101.
[PMID: 1521099] ]. In addition to HSCT and BMT, another group of physicians reported the successful use of umbilical cord blood (UCB)-derived stem cells in a 3-month-old patient who had lived to age 3 ½ at the time of publication [71Stein J, Garty BZ, Dror Y, Fenig E, Zeigler M, Yaniv I. Successful treatment of Wolman disease by unrelated umbilical cord blood transplantation. Eur J Pediatr 2007; 166(7): 663-6.
[http://dx.doi.org/10.1007/s00431-006-0298-6] [PMID: 17033804] ]. The patient’s severe hepatosplenomegaly completely vanished, but the adrenal glands remained calcified. The patient did not show any signs of adrenal insufficiency and was growing normally. Nevertheless, HSCT or BMT remains a dangerous procedure with many possible complications.
Unlike WD, liver transplants in patients with CESD have proven successful. Given that the disease pathology that results in premature demise is cirrhosis of the liver, transplantation is a logical medical procedure to extend the life of the patient. Reports in the literature confirm patient survival ranging from 2 years post-operation to 7 years [82Arterburn JN, Lee WM, Wood RP, Shaw BW, Markin RS. Orthotopic liver transplantation for cholesteryl ester storage disease. J Clin Gastroenterol 1991; 13(4): 482-5.
[http://dx.doi.org/10.1097/00004836-199108000-00028] [PMID: 1918862] , 83Ferry GD, Whisennand HH, Finegold MJ, Alpert E, Glombicki A. Liver transplantation for cholesteryl ester storage disease. J Pediatr Gastroenterol Nutr 1991; 12(3): 376-8.
[http://dx.doi.org/10.1097/00005176-199104000-00016] [PMID: 2072231] ]. After transplantation of a healthy liver into a 10-year-old patient, Arterburn et al. (1991) noted that the patient continued to have full activity, normal growth and development, and normal liver function tests. The patient’s own (excised) liver was examined and was found to be grossly cirrhotic. Liver transplantation, however, remains a risky procedure and is not a permanent solution. Kale et al. (1995) reported on a 21-year-old female who developed end-stage renal failure after a successful liver transplant surgery 7 years prior. The use of nephrotoxic agents for immunosuppression post-op may have contributed to the deterioration of the patient’s kidneys [84Kale AS, Ferry GD, Hawkins EP. End-stage renal disease in a patient with cholesteryl ester storage disease following successful liver transplantation and cyclosporine immunosuppression. J Pediatr Gastroenterol Nutr 1995; 20(1): 95-7.
[http://dx.doi.org/10.1097/00005176-199501000-00016] [PMID: 7884624] ].
Enzyme Replacement Therapy
Several other lysosomal storage diseases, such as Gaucher disease, have been treated with enzyme replacement therapy (ERT) [85Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiencymacrophage-targeted glucocerebrosidase for Gauchers disease. N Engl J Med 1991; 324(21): 1464-70.
[http://dx.doi.org/10.1056/NEJM199105233242104] [PMID: 2023606] ]. In this strategy, recombinant proteins of a lysosomal enzyme absent in the LSD are produced and then delivered to patients intravenously. These recombinant enzymes are endocytosed into cells via the mannose-6-phosphate receptor [86Braulke T, Bonifacino J. Sorting of lysosomal proteins. Biochim et Biophys Acta 2009; 1793(4): 605-14.
[http://dx.doi.org/10.1016/j.bbamcr.2008.10.016] ]. Among the six LSDs that have been treated with ERT (Fabry, MPS I, II, and VI, and glycogen storage type II/Pompe), Gaucher disease has been by far the most successful [87Lachmann RH. Enzyme replacement therapy for lysosomal storage diseases. Curr Opin Pediatr 2011; 23(6): 588-93.
[http://dx.doi.org/10.1097/MOP.0b013e32834c20d9] [PMID: 21946346] ]. This success is most likely due to the efficient delivery of the recombinant enzyme to target tissues, which happen to be tissues highly enriched with blood vessels and thus able to effectively receive sufficient amounts of these enzymes. Since enzymes do not cross the blood brain barrier, ERT is not effective in treating LSDs with neurological symptoms. ERT in CESD was first examined in vitro using cultured CESD fibroblasts, which took up exogenously produced LAL [88Poznansky MJ, Hutchison SK, Davis PJ. Enzyme replacement therapy in fibroblasts from a patient with cholesteryl ester storage disease. FASEB J 1989; 3(2): 152-6.
[PMID: 2644147] ]. The enzyme, bound to albumin as a stabilization agent, decreased levels of cholesteryl esters by half. WD fibroblasts can take up human LAL in vitro, with subsequent degradation of intracellularly accumulated CE [89Sando GN, Henke VL. Recognition and receptor-mediated endocytosis of the lysosomal acid lipase secreted by cultured human fibroblasts. J Lipid Res 1982; 23(1): 114-23.
[PMID: 7057100] ]. In a LAL-/- mouse model of WD, human recombinant LAL was well-tolerated and ameliorated several symptoms of the disease [28Du H, Schiavi S, Levine M, Mishra J, Heur M, Grabowski GA. Enzyme therapy for lysosomal acid lipase deficiency in the mouse. Hum Mol Genet 2001; 10(16): 1639-48.
[http://dx.doi.org/10.1093/hmg/10.16.1639] [PMID: 11487567] ]. In order to test if ERT was a viable therapy for WD and CESD in the LAL mouse model, mice were treated via tail vein injections and showed a reduction in hepatic yellow coloration and overall weight, in addition to a reduction in the number and size of Kupffer cells and tissue- infiltrating macrophages. In addition, TG and CE levels were decreased by 50% in the liver, 69% in the spleen, and 50% in the small intestine. Given that exogenously produced enzyme delivered intravenously tends to accumulate in the spleen, liver, and bone marrow, it is reasonable to believe that ERT would show some success in WD patients [90Mistry PK, Wraight EP, Cox TM. Therapeutic delivery of proteins to macrophages: implications for treatment of Gauchers disease. Lancet 1996; 348(9041): 1555-9.
[http://dx.doi.org/10.1016/S0140-6736(96)04451-0] [PMID: 8950883] ]. A phase 3 clinical trial using sebelipase alfa (Kanuma™) as ERT in WD and CESD was initiated in 2012 by Synageva BioPharma and has resulted in a recent approval by the FDA [91FDA approves first drug to treat a rare enzyme disorder in pediatric and adult patients. US Department of Health and Human Services: USA. 2015.]. In a double-blind, placebo controlled study involving 66 patients with CESD, sebilipase alfa treatment resulted in higher rates of normalization of the alanine aminotransferase level compared to placebo. Additionally, treatment resulted in significant changes in six other secondary endpoints including a change from baseline in LDL cholesterol level, non-HDL cholesterol level, aspartate aminotransferase, triglyceride levels, HDL cholesterol levels, and hepatic fat content [92Burton BK, Balwani M, Feillet F, et al. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med 2015; 373(11): 1010-20.
[http://dx.doi.org/10.1056/NEJMoa1501365] [PMID: 26352813] ]. In an open-label study of nine patients, seblipase alfa was well tolerated and resulted in improvements to key serum disease activity markers, including alanine and aspartate transaminases. Patients had improved weight gain, reduced abdominal distention, hepatosplenomegaly and a resolution of vomiting and diarrhea. Additionally, six of the nine patients were alive at twelve months of age compared to none of the twenty-one historical controls [93Valyannopoulos V, Plantaz D, Vara R, et al. Clinical effect of sebelipase alfa on survival and growth in infants with lysosomal acid lipase deficiency (Wolman disease). Mol Genet Metab 2014; 111: S108.
[http://dx.doi.org/10.1016/j.ymgme.2013.12.268] ]. This is the first treatment for patients with LAL deficiency.
Small Molecule Drugs
While no small molecule treatments are approved for WD, statins have been used to treat CESD. Statins reduce cholesterol synthesis by inhibition of 3-hydroxy-3-methylglutaryl-coenzymeA reductase (HMG-CoA), which catalyzes an important step in hepatic cholesterol synthesis [94Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation 2000; 101(2): 207-13.
[http://dx.doi.org/10.1161/01.CIR.101.2.207] [PMID: 10637210] ]. Lovastatin and simvastatin have both been utilized in the clinic with varying rates of success. In some cases, statin therapy reduced hepatosplenomegaly and TG/CE levels in serum and in the liver [74Gasche C, Aslanidis C, Kain R, et al. A novel variant of lysosomal acid lipase in cholesteryl ester storage disease associated with mild phenotype and improvement on lovastatin. J Hepatol 1997; 27(4): 744-50.
[http://dx.doi.org/10.1016/S0168-8278(97)80092-X] [PMID: 9365051] , 95Ginsberg HN, Le NA, Short MP, Ramakrishnan R, Desnick RJ. Suppression of apolipoprotein B production during treatment of cholesteryl ester storage disease with lovastatin. Implications for regulation of apolipoprotein B synthesis. J Clin Invest 1987; 80(6): 1692-7.
[http://dx.doi.org/10.1172/JCI113259] [PMID: 3680522] -97Tarantino MD, McNamara DJ, Granstrom P, Ellefson RD, Unger EC, Udall JN Jr. Lovastatin therapy for cholesterol ester storage disease in two sisters. J Pediatr 1991; 118(1): 131-5.
[http://dx.doi.org/10.1016/S0022-3476(05)81866-9] [PMID: 1986080] ] while in other cases the reduction in TG/CE levels was not significant [98Glueck CJ, Lichtenstein P, Tracy T, Speirs J. Safety and efficacy of treatment of pediatric cholesteryl ester storage disease with lovastatin. Pediatr Res 1992; 32(5): 559-65.
[http://dx.doi.org/10.1203/00006450-199211000-00015] [PMID: 1480458] , 99Di Bisceglie AM, Ishak KG, Rabin L, Hoeg JM. Cholesteryl ester storage disease: hepatopathology and effects of therapy with lovastatin. Hepatology 1990; 11(5): 764-72.
[http://dx.doi.org/10.1002/hep.1840110509] [PMID: 2347551] ]. The efficacy of statins is augmented when used in combination with other cholesterol lowering drugs, such as ezetimibe [100Tadiboyina VT, Liu DM, Miskie BA, Wang J, Hegele RA. Treatment of dyslipidemia with lovastatin and ezetimibe in an adolescent with cholesterol ester storage disease. Lipids Health Dis 2005; 4: 26-32.
[http://dx.doi.org/10.1186/1476-511X-4-26] [PMID: 16255772] ] and cholestyramine [101Yokoyama S, McCoy E. Long-term treatment of a homozygous cholesteryl ester storage disease with combined cholestyramine and lovastatin. J Inherit Metab Dis 1992; 15(2): 291-2.
[http://dx.doi.org/10.1007/BF01799650] [PMID: 1528002] ]. The varied success rates in using statins to-treat CESD patients are likely due to the heterogeneity of clinical severity found with CESD. Animal studies have shown that statins not only reduce cholesterol synthesis, but also increase the hepatic uptake of LDL through the upregulation of LDL receptor activity. Given that CESD and WD cause a disease phenotype as a result of the over-accumulation of TG and CEs in the lysosome from LDL, it may seem counterintuitive that this therapy would work in patients. However, it has been hypothesized that if the overall reduction in native cholesterol biosynthesis in the liver is greater than upregulation of LDL receptor activity due to statin treatment, the net benefit of statin treatment would be reduced storage of TGs and CEs [102Levy R, Ostlund RE Jr, Schonfeld G, Wong P, Semenkovich CF. Cholesteryl ester storage disease: complex molecular effects of chronic lovastatin therapy. J Lipid Res 1992; 33(7): 1005-15.
[PMID: 1431581] , 103Iverson SA, Cairns SR, Ward CP, Fensom AH. Asymptomatic cholesteryl ester storage disease in an adult controlled with simvastatin. Ann Clin Biochem 1997; 34(Pt 4): 433-6.
[http://dx.doi.org/10.1177/000456329703400418] [PMID: 9247680] ]. Statins and other cholesterol lowering drugs have not been used to treat WD. Due to the severity of the disease phenotype of WD compared to CESD and the long-term treatment regimens (>1 year), it is likely that statins would not be very efficacious for WD.
While Kanuma represents a critical step forward in the therapeutic development for LAL deficiencies, a small molecule therapy would be an excellent asset for a holistic treatment strategy. High throughput drug screening (HTS) of chemical libraries is a potential method for rapid identification of active compounds (“hits”) that ameliorate disease phenotypes in patient-derived cells [104Macarron R, Banks MN, Bojanic D, et al. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov 2011; 10(3): 188-95.
[http://dx.doi.org/10.1038/nrd3368] [PMID: 21358738] ]. Phenotypic screening is particularly amenable to LSDs, as disease cells such as skin fibroblasts and blood cells that usually exhibit characteristic disease phenotypes can be obtained from patients. Recent advances in high-content cellular imaging technology allows rapid compound screens using phenotypic assays. Novel lead compounds that reduce accumulated macromolecules in the lysosome or inhibit pathways that synthesize the accumulating substrates may be identified using this method [105Huang H-P, Chuang C-Y, Kuo H-C. Induced pluripotent stem cell technology for disease modeling and drug screening with emphasis on lysosomal storage diseases. Stem Cell Res Ther 2012; 3(4): 34.
[http://dx.doi.org/10.1186/scrt125] [PMID: 22925465] ]. The LysoTracker dye staining assay has been reported to be a good assay for measuring enlarged lysosomes in WD cells [106Xu M, Liu K, Swaroop M, et al. A phenotypic compound screening assay for lysosomal storage diseases. J Biomol Screen 2014; 19(1): 168-75.
[http://dx.doi.org/10.1177/1087057113501197] [PMID: 23983233] ]. Accumulation of nonpolar lipids was also detected in the Nile-Red dye staining assay using WD cells (Fig. 1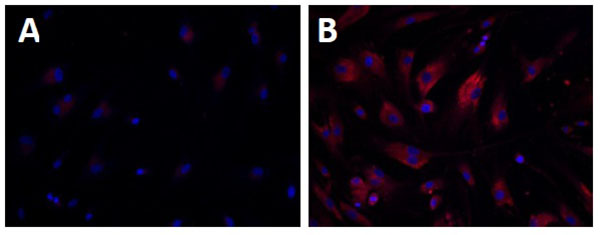 ) [107Xu M, Liu K, Swaroop M, et al. δ-Tocopherol reduces lipid accumulation in Niemann-Pick type C1 and Wolman cholesterol storage disorders. J Biol Chem 2012; 287(47): 39349-60.
) [107Xu M, Liu K, Swaroop M, et al. δ-Tocopherol reduces lipid accumulation in Niemann-Pick type C1 and Wolman cholesterol storage disorders. J Biol Chem 2012; 287(47): 39349-60.
[http://dx.doi.org/10.1074/jbc.M112.357707] [PMID: 23035117] ]. Both these assays can be used for high throughput screening of compound collections to identify lead compounds for drug development to treat WD and CESD.
WD and CESD Disease Models
In order to develop novel therapeutics directed to act on WD and CESD, disease models that recapitulate the disease pathology and phenotype are required. In the late 1990’s, a WD LIPA knockout mouse model was generated [108Du H, Duanmu M, Witte D, Grabowski GA. Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Hum Mol Genet 1998; 7(9): 1347-54.
[http://dx.doi.org/10.1093/hmg/7.9.1347] [PMID: 9700186] ]. Recently, induced pluripotent stem cell (iPSC) technology has been used to generate patient-derived and disease-relevant cellular models for other lysosomal storage diseases [109Lemonnier T, Blanchard S, Toli D, et al. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum Mol Genet 2011; 20(18): 3653-66.
[http://dx.doi.org/10.1093/hmg/ddr285] [PMID: 21685203] -111Yu D, Swaroop M, Wang M, et al. Niemann-pick disease type C: induced pluripotent stem cell-derived neuronal cells for modeling neural disease and evaluating drug efficacy. J Biomol Screen 2014; 19(8): 1164-73.
[http://dx.doi.org/10.1177/1087057114537378] [PMID: 24907126] ]. While it has not been reported for WD or CESD, an appropriate cell-based disease model derived from patient iPSCs would provide a useful tool for small molecule drug screening.

Animal Models
Traditionally, animal models have been used for both the study of disease pathophysiology and evaluation of drug efficacy. The results from such models provide important insight into the pathology of diseases in vivo. In 1998, the first animal model of WD and CESD, a LIPA homozygous knockout mouse, was generated [108Du H, Duanmu M, Witte D, Grabowski GA. Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Hum Mol Genet 1998; 7(9): 1347-54.
[http://dx.doi.org/10.1093/hmg/7.9.1347] [PMID: 9700186] ]. The only apparent presentation of these mice was a lower than normal weight. These mice had orange-colored livers and slightly enlarged spleens. However, lipid analyses from the liver, spleen, and small intestine show massive amounts of accumulated TG and CE, with liver CEs at ~35 fold higher compared to wild-type mice. A histological analysis found enlarged hepatocytes, Kupffer cells, adrenal glands, and intestinal villi. Adrenal calcification did not occur in this mouse model. Although mice lived into early adulthood, death occurred due to hepatosplenomegaly and malabsorption. Because of its survival into adulthood, as well as intestinal pathology similar to CESD, this model can be used to investigate both diseases on the LAL deficiency spectrum. The liver pathology in this model is very similar to WD, and while adrenal calcification does not occur, the adrenal gland is still histologically similar to WD in humans [99Di Bisceglie AM, Ishak KG, Rabin L, Hoeg JM. Cholesteryl ester storage disease: hepatopathology and effects of therapy with lovastatin. Hepatology 1990; 11(5): 764-72.
[http://dx.doi.org/10.1002/hep.1840110509] [PMID: 2347551] ] Interestingly, a naturally occurring rat model of WD, which is also LIPA null, displayed a similar phenotype to the murine model, without presenting any adrenal calcification [112Kuriyama M, Yoshida H, Suzuki M, Fujiyama J, Igata A. Lysosomal acid lipase deficiency in rats: lipid analyses and lipase activities in liver and spleen. J Lipid Res 1990; 31(9): 1605-12.
[PMID: 2246613] ]. While these two model animals may recapitulate many aspects of the WD phenotype, significant enough differences have been found between these rodent models and human patients to warrant the generation of more human disease relevant models of WD. Given that mutations in CESD allow for some production of functional LAL (3-8%), animal models of CESD should be created that possess mutations representative of this disease.
Potential for “Disease-in-a-Dish” Models of LAL Deficiency
Since the first iPSC was generated from adult cells by Yamanaka and colleagues [113Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126(4): 663-76.
[http://dx.doi.org/10.1016/j.cell.2006.07.024] [PMID: 16904174] ], the methods for iPSC generation and mature cell differentiation have been rapidly developed. While iPSCs and their derivative cells have potential for direct clinical applications for the treatment of various diseases, patient derived iPSCs can be differentiated into mature cell types such as neurons, cardiomyocytes and hepatocytes for modeling disease phenotypes [110Zhang J, Lian Q, Zhu G, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011; 8(1): 31-45.
[http://dx.doi.org/10.1016/j.stem.2010.12.002] [PMID: 21185252] , 114Dimos JT, Rodolfa KT, Niakan KK, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008; 321(5893): 1218-21.
[http://dx.doi.org/10.1126/science.1158799] [PMID: 18669821] ]. Drug screening campaigns conducted on these cells would be an alternative approach for drug development [115Spence J, Mayhew C, Rankin S, et al. Directed differentiation of human pluripotent stem cells into intestinal tissues in vitro. Nature 2011; 470(7332): 105-9.
[http://dx.doi.org/10.1038/nature09691] , 116Chen Y-F, Tseng C-Y, Wang H-W, Kuo H-C, Yang VW, Lee OK. Rapid generation of mature hepatocyte-like cells from human induced pluripotent stem cells by an efficient three-step protocol. Hepatology 2012; 55(4): 1193-203.
[http://dx.doi.org/10.1002/hep.24790] [PMID: 22095466] ].
Several iPSC lines have been reported for lysosomal storage diseases using iPSCs. Murine iPSCs were generated from mouse models of Fabry disease, globoid cell leukodystrophy (GLD), and mucopolysaccharidosis VII (MPSVII) [117Meng X-L, Shen J-S, Kawagoe S, Ohashi T, Brady RO, Eto Y. Induced pluripotent stem cells derived from mouse models of lysosomal storage disorders. Proc Natl Acad Sci USA 2010; 107(17): 7886-91.
[http://dx.doi.org/10.1073/pnas.1002758107] [PMID: 20385825] ]. The iPSCs from the Fabry disease mice showed high immunostaining for globotriaosylceramide, a hallmark of the disease. The iPSCs differentiated cardiomyocytes, a major cell type affected by Fabry disease, also showed the same pattern of globotriaosylceramide accumulation. iPSCs generated from GLD- and MPSVII-affected mice also demonstrated appropriate defects, proving that murine-derived iPSCs strongly model their respective LSDs.
iPSCs have also been generated for mucopolysaccharidosis type IIIB (MPSVIIIB) from patient fibroblasts, a fatal lysosomal storage disease that induces pathology primarily in CNS. The abnormality in Golgi apparatus was observed in both patient iPSCs and differentiated neural stem cells [109Lemonnier T, Blanchard S, Toli D, et al. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum Mol Genet 2011; 20(18): 3653-66.
[http://dx.doi.org/10.1093/hmg/ddr285] [PMID: 21685203] ]. For Pompe disease, Huang et al. (2011) reported that cardiomyocytes derived from Pompe iPSCs recapitulated the disease phenotypes, including low acid alpha-glucosidase activity, glycogen accumulation and multiple ultrastructural aberrances [118Huang H-P, Chen P-H, Hwu W-L, et al. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum Mol Genet 2011; 20(24): 4851-64.
[http://dx.doi.org/10.1093/hmg/ddr424] [PMID: 21926084] ]. These cells were responsive to drug treatment or affect cellular processes relevant to Pompe disease, demonstrating that patient iPSC-derived cells can serve as a drug discovery platform for Pompe disease. The iPSC derived cell-based diseased models were also reported for Niemann-Pick Disease Type C (NPC), another lysosomal storage disease [111Yu D, Swaroop M, Wang M, et al. Niemann-pick disease type C: induced pluripotent stem cell-derived neuronal cells for modeling neural disease and evaluating drug efficacy. J Biomol Screen 2014; 19(8): 1164-73.
[http://dx.doi.org/10.1177/1087057114537378] [PMID: 24907126] , 119Maetzel D, Sarkar S, Wang H, et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep 2014; 2(6): 866-80.
[http://dx.doi.org/10.1016/j.stemcr.2014.03.014] [PMID: 24936472] , 120Swaroop M, Thorne N, Rao MS, Austin CP, McKew JC, Zheng W. Evaluation of cholesterol reduction activity of methyl-β-cyclodextrin using differentiated human neurons and astrocytes. J Biomol Screen 2012; 17(9): 1243-51.
[http://dx.doi.org/10.1177/1087057112456877] [PMID: 22923786] ]. Maetzal et al. (2014) generated iPSCs and demonstrated that these cells have impaired cholesterol metabolism and a defect in autophagy [119Maetzel D, Sarkar S, Wang H, et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep 2014; 2(6): 866-80.
[http://dx.doi.org/10.1016/j.stemcr.2014.03.014] [PMID: 24936472] ]. Another report showed that treatment of iPSC derived neuronal cells with delta-tocopherol, hydroxypropyl-beta-cyclodextrin, and methyl-beta-cyclodextrin showed a significant reduction in lysosomal storage of unesterified cholesterol [111Yu D, Swaroop M, Wang M, et al. Niemann-pick disease type C: induced pluripotent stem cell-derived neuronal cells for modeling neural disease and evaluating drug efficacy. J Biomol Screen 2014; 19(8): 1164-73.
[http://dx.doi.org/10.1177/1087057114537378] [PMID: 24907126] , 120Swaroop M, Thorne N, Rao MS, Austin CP, McKew JC, Zheng W. Evaluation of cholesterol reduction activity of methyl-β-cyclodextrin using differentiated human neurons and astrocytes. J Biomol Screen 2012; 17(9): 1243-51.
[http://dx.doi.org/10.1177/1087057112456877] [PMID: 22923786] ].
Patient iPSC derived disease models are advantageous as they can generate disease relevant cells and can accurately capture a patient’s genetic background. This approach allows drug screening in patient cells with varying mutations and disease phenotypes. For WD and CESD, compound screening using patient-iPSC derived disease models may lead to identification of three types of lead compounds. The first type of active compounds may reduce the amount of CEs and TGs entering lysosomes, as statins have shown to alleviate symptoms in CESD patients via the inhibition of cholesterol synthesis. Miglustat, approved for use in patients with Gaucher type I and NPC, is an example of enzyme reduction therapy [121Smid BE, Aerts JM, Boot RG, Linthorst GE, Hollak CE. Pharmacological small molecules for the treatment of lysosomal storage disorders. Expert Opin Investig Drugs 2010; 19(11): 1367-79.
[http://dx.doi.org/10.1517/13543784.2010.524205] [PMID: 20942596] ], as well as Eliglustat for Gaucher type 1 [122Mistry PK, Lukina E, Ben Turkia H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA 2015; 313(7): 695-706.
[http://dx.doi.org/10.1001/jama.2015.459] [PMID: 25688781] ]. A second type of active compound may stabilize misfolded proteins that result from missense mutations of the LIPA gene. Misfolded proteins are retained by the endoplasmic reticulum associated degradation system and then degraded in proteasome [123Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 2003; 4(3): 181-91.
[http://dx.doi.org/10.1038/nrm1052] [PMID: 12612637] ]. Small molecules may bind to the mutant enzyme proteins, resulting in increased enzyme stability and allowing them to avoid the ERAD system for degradation and to be properly trafficking to the lysosome [124Valenzano KJ, Khanna R, Powe AC, et al. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay Drug Dev Technol 2011; 9(3): 213-35.
[http://dx.doi.org/10.1089/adt.2011.0370] [PMID: 21612550] ]. The third type of active compound may have the ability to reduce the accumulated CEs and TGs by other mechanisms. For example, cyclodextrins have been found to remove accumulated unesterified cholesterol from NPC cells as well as in the NPC mouse model by induction of autophagy [125Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2011; 8(2): 108-17.
[http://dx.doi.org/10.1038/nrneurol.2011.200] [PMID: 22187000] ]. Targeting the autophagy pathway could be useful for therapeutic development in treating LSDs [126].
Compared with ERT, small-molecule based therapies hold several advantages. First, small molecule drugs can be taken orally by patients, whereas ERT requires intravenous administration during lengthy clinic visits. Second, small molecule drugs can be optimized to penetrate into various tissues including CNS, while ERT is only useful for treatments of peripheral symptoms. Lastly, with an average of $90,000 to $720,000 per year for ERT per patient, the astronomically high cost of ERT is a tremendous burden for patients [127Wraith JE. Limitations of enzyme replacement therapy: current and future. J Inherit Metab Dis 2006; 29(2-3): 442-7.
[http://dx.doi.org/10.1007/s10545-006-0239-6] [PMID: 16763916] ]. In addition, a combination of both small molecule therapy and ERT could be synergistic, thus reducing the need of frequent dosing for ERT and improving the therapeutic effects.
CONCLUSION
WD and CESD are rare genetic diseases caused by mutations in the LIPA gene. The variable symptoms associated with CESD cause underdiagnoses of this disease. HSCT and BMT are associated with an unacceptably high mortality rate and their therapeutic outcome is also uncertain. ERT has recently been approved by the FDA for the treatment of patients with LAL-deficiency. Similar to other LSDs, small molecule drugs are still needed for future improved of treatment of WD and CESD. Recent advances in iPSC technology have provided a new cell based disease model system for WD and CESD with patient derived cells. High throughput screening of compound collections with phenotypic disease models of WD and CESD developed by iPSC technology will enable the development of new small molecule drugs for treatments of both diseases (Fig. 2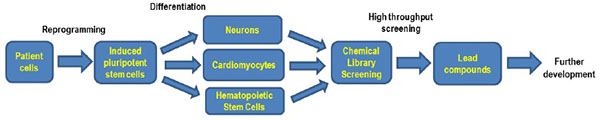 ).
).
 |
Fig. (2) Flowchart of iPS cells based drug development. |
AUTHORS CONTRIBUTIONS
FA performed the literature search and wrote the manuscript. NT and WZ provided critical edits and revisions to the manuscript. All authors read and approved the final manuscript.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
This work was supported by the Intramural Research Program of the National Center for Advancing Translational Sciences, the National Institutes of Health.
REFERENCES
| [1] | Platt F, Boland B, van der Spoel A. Lysosomal storage disorders: the cellular impact of lysosomal function. J Cell Biol 2012; 199: 723-34. [http://dx.doi.org/10.1083/jcb.201208152] [PMID: 23185029] |
| [2] | Xu M, Motabar O, Ferrer M, Marugan JJ, Zheng W, Ottinger EA. Disease models for the development of therapies for lysosomal storage diseases. Ann N Y Acad Sci 2016; 1371(1): 15-29. [http://dx.doi.org/10.1111/nyas.13052] [PMID: 27144735] |
| [3] | Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis 2010; 5: 16. [http://dx.doi.org/10.1186/1750-1172-5-16] [PMID: 20525256] |
| [4] | Toscano A, Schoser B. Enzyme replacement therapy in late-onset Pompe disease: a systematic literature review. J Neurol 2013; 260(4): 951-9. [http://dx.doi.org/10.1007/s00415-012-6636-x] [PMID: 22926164] |
| [5] | Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999; 281(3): 249-54. [http://dx.doi.org/10.1001/jama.281.3.249] [PMID: 9918480] |
| [6] | Brady RO. Benefits from unearthing a biochemical Rosetta Stone. J Biol Chem 2010; 285(53): 41216-21. [http://dx.doi.org/10.1074/jbc.X110.197954] [PMID: 21030589] |
| [7] | Pastores GM. Miglustat: substrate reduction therapy for lysosomal storage disorders associated with primary central nervous system involvement. Recent Patents CNS Drug Discov 2006; 1(1): 77-82. [http://dx.doi.org/10.2174/157488906775245282] [PMID: 18221193] |
| [8] | Abramov A, Schorr S, Wolman M. Generalized xanthomatosis with calcified adrenals. AMA J Dis Child 1956; 91(3): 282-6. [PMID: 13301142] |
| [9] | Young EP, Patrick AD. Deficiency of acid esterase activity in Wolmans disease. Arch Dis Child 1970; 45(243): 664-8. [http://dx.doi.org/10.1136/adc.45.243.664] [PMID: 5477680] |
| [10] | Maehira F, Nakada F, Hokama T. Characteristics of acid esterase in Wolmans disease. Biochem Med 1984; 32(3): 322-30. [http://dx.doi.org/10.1016/0006-2944(84)90037-1] [PMID: 6517876] |
| [11] | Porto AF. Lysosomal acid lipase deficiency: diagnosis and treatment of Wolman and Cholesteryl Ester Storage Diseases. Pediatr Endocrinol Rev 2014; 12(Suppl. 1): 125-32. [PMID: 25345094] |
| [12] | Wolman M. Wolman disease and its treatment. Clin Pediatr (Phila) 1995; 34(4): 207-12. [http://dx.doi.org/10.1177/000992289503400406] [PMID: 7789014] |
| [13] | Wolman Disease. Genetics Home Reference Available at: https://ghrnlmnihgov/condition/wolman-disease 2007. |
| [14] | Valles-Ayoub Y, Esfandiarifard S, No D, et al. Wolman disease (LIPA p.G87V) genotype frequency in people of Iranian-Jewish ancestry. Genet Test Mol Biomarkers 2011; 15(6): 395-8. [http://dx.doi.org/10.1089/gtmb.2010.0203] [PMID: 21291321] |
| [15] | Muntoni S, Wiebusch H, Jansen-Rust M, et al. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol 2007; 27(8): 1866-8. [http://dx.doi.org/10.1161/ATVBAHA.107.146639] [PMID: 17634524] |
| [16] | Bernstein DL, Hülkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 2013; 58(6): 1230-43. [http://dx.doi.org/10.1016/j.jhep.2013.02.014] [PMID: 23485521] |
| [17] | Burton BK, Deegan PB, Enns GM, et al. Clinical features of lysosomal acid lipase deficiency. J Pediatr Gastroenterol Nutr 2015; 61(6): 619-25. [http://dx.doi.org/10.1097/MPG.0000000000000935] [PMID: 26252914] |
| [18] | Coates PM, Cortner JA, Mennuti MT, Wheeler JE. Prenatal diagnosis of Wolman disease. Am J Med Genet 1978; 2(4): 397-407. [http://dx.doi.org/10.1002/ajmg.1320020409] [PMID: 122435] |
| [19] | Patrick AD, Willcox P, Stephens R, Kenyon VG. Prenatal diagnosis of Wolmans disease. J Med Genet 1976; 13(1): 49-51. [http://dx.doi.org/10.1136/jmg.13.1.49] [PMID: 1271426] |
| [20] | Shome DK, Al-Jishi E, Greally JF, Malik N, Zainaldeen HA, Das NS. The Middle-East connection of Wolman Disease. Saudi Med J 2002; 23(5): 597-601. [PMID: 12070591] |
| [21] | Crocker AC, Vawter GF, Neuhauser EB, Rosowsky A. Wolmans disease: three new patients with a recently described lipidosis. Pediatrics 1965; 35: 627-40. [PMID: 14269714] |
| [22] | Low G, Irwin G, MacPhee G, Robinson P. Characteristic imaging findings in wolman's disease. Clin Radiol Extra 2004; 59: 106-8. [http://dx.doi.org/10.1016/j.cradex.2004.05.002] |
| [23] | Sadhukhan M, Saha A, Vara R, Bhaduri B. Infant case of lysosomal acid lipase deficiency: Wolmans disease. BMJ Case Rep 2014; 2014: 1-3. [http://dx.doi.org/10.1136/bcr-2013-202652] [PMID: 24832708] |
| [24] | Shenoy P, Karegowda L, Sripathi S, Mohammed N. Wolman disease in an infant. In: BMJ Case Reports. 2014; 2014. pii: bcr2014203656. |
| [25] | Queloz JM, Capitanio MA, Kirkpatrick JA. Wolmans disease. Roentgen observations in 3 siblings. Radiology 1972; 104(2): 357-9. [http://dx.doi.org/10.1148/104.2.357] [PMID: 5044990] |
| [26] | Schaub J, Janka GE, Christomanou H, et al. Wolmans disease: clinical, biochemical and ultrastructural studies in an unusual case without striking adrenal calcification. Eur J Pediatr 1980; 135(1): 45-53. [http://dx.doi.org/10.1007/BF00445892] [PMID: 7449788] |
| [27] | Hill SC, Hoeg JM, Dwyer AJ, Vucich JJ, Doppman JL. CT findings in acid lipase deficiency: wolman disease and cholesteryl ester storage disease. J Comput Assist Tomogr 1983; 7(5): 815-8. [http://dx.doi.org/10.1097/00004728-198310000-00011] [PMID: 6886132] |
| [28] | Du H, Schiavi S, Levine M, Mishra J, Heur M, Grabowski GA. Enzyme therapy for lysosomal acid lipase deficiency in the mouse. Hum Mol Genet 2001; 10(16): 1639-48. [http://dx.doi.org/10.1093/hmg/10.16.1639] [PMID: 11487567] |
| [29] | Lake BD, Patrick AD. Wolmans disease: deficiency of E600-resistant acid esterase activity with storage of lipids in lysosomes. J Pediatr 1970; 76(2): 262-6. [http://dx.doi.org/10.1016/S0022-3476(70)80172-X] [PMID: 5410174] |
| [30] | Boldrini R, Devito R, Biselli R, Filocamo M, Bosman C. Wolman disease and cholesteryl ester storage disease diagnosed by histological and ultrastructural examination of intestinal and liver biopsy. Pathol Res Pract 2004; 200(3): 231-40. [http://dx.doi.org/10.1016/j.prp.2003.11.001] [PMID: 15200275] |
| [31] | Wolman M, Sterk VV, Gatt S, Frenkel M. Primary familial xanthomatosis with involvement and calcification of the adrenals. Report of two more cases in siblings of a previously described infant. Pediatrics 1961; 28: 742-57. [PMID: 14008104] |
| [32] | Röyttä M, Fagerlund AS, Toikkanen S, et al. Wolman disease: morphological, clinical and genetic studies on the first Scandinavian cases. Clin Genet 1992; 42(1): 1-7. [http://dx.doi.org/10.1111/j.1399-0004.1992.tb03125.x] [PMID: 1516222] |
| [33] | Kikuchi M, Igarashi K, Noro T, Igarashi Y, Hirooka M, Tada K. Evaluation of jejunal function in Wolmans disease. J Pediatr Gastroenterol Nutr 1991; 12(1): 65-9. [http://dx.doi.org/10.1097/00005176-199101000-00014] [PMID: 2061782] |
| [34] | Grabowski G, Du H, Charnas L. Lysosomal acid lipase deficiencies: the wolman disease/cholesteryl ester storage disease spectrum the online metabolic & molecular bases of inherited disease. New York: McGraw-Hill 2013; p. 142. |
| [35] | Chatrath H, Keilin S, Attar BM. Cholesterol ester storage disease (CESD) diagnosed in an asymptomatic adult. Dig Dis Sci 2009; 54(1): 168-73. [http://dx.doi.org/10.1007/s10620-008-0310-2] [PMID: 18478331] |
| [36] | Hůlková H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology 2012; 60(7): 1107-13. [http://dx.doi.org/10.1111/j.1365-2559.2011.04164.x] [PMID: 22621222] |
| [37] | Kwiterovich PO Jr. The metabolic pathways of high-density lipoprotein, low-density lipoprotein, and triglycerides: a current review. Am J Cardiol 2000; 86(12A): 5L-10L. [http://dx.doi.org/10.1016/S0002-9149(00)01461-2] [PMID: 11374859] |
| [38] | Drebber U, Andersen M, Kasper HU, Lohse P, Stolte M, Dienes HP. Severe chronic diarrhea and weight loss in cholesteryl ester storage disease: a case report. World J Gastroenterol 2005; 11(15): 2364-6. [http://dx.doi.org/10.3748/wjg.v11.i15.2364] [PMID: 15818756] |
| [39] | Groener JE, Bax W, Stuani C, Pagani F. Difference in substrate specificity between human and mouse lysosomal acid lipase: low affinity for cholesteryl ester in mouse lysosomal acid lipase. Biochim Biophys Acta 2000; 1487(2-3): 155-62. [http://dx.doi.org/10.1016/S1388-1981(00)00091-3] [PMID: 11018468] |
| [40] | Beaudet AL, Ferry GD, Nichols BL Jr, Rosenberg HS. Cholesterol ester storage disease: clinical, biochemical, and pathological studies. J Pediatr 1977; 90(6): 910-4. [http://dx.doi.org/10.1016/S0022-3476(77)80557-X] [PMID: 859064] |
| [41] | Edelstein RA, Filling-Katz MR, Pentchev P, et al. Cholesteryl ester storage disease: a patient with massive splenomegaly and splenic abscess. Am J Gastroenterol 1988; 83(6): 687-92. [PMID: 3376925] |
| [42] | Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta 2012; 413(15-16): 1207-10. [http://dx.doi.org/10.1016/j.cca.2012.03.019] [PMID: 22483793] |
| [43] | Dairaku T, Iwamoto T, Nishimura M, Endo M, Ohashi T, Eto Y. A practical fluorometric assay method to measure lysosomal acid lipase activity in dried blood spots for the screening of cholesteryl ester storage disease and Wolman disease. Mol Genet Metab 2014; 111(2): 193-6. [http://dx.doi.org/10.1016/j.ymgme.2013.11.003] [PMID: 24295952] |
| [44] | Christomanou H, Cáp C. Prenatal monitoring for Wolmans disease in a pregnancy at risk: first case in the Federal Republic of Germany. Hum Genet 1981; 57(4): 440-1. [http://dx.doi.org/10.1007/BF00281702] [PMID: 7286988] |
| [45] | Desai PK, Astrin KH, Thung SN, et al. Cholesteryl ester storage disease: pathologic changes in an affected fetus. Am J Med Genet 1987; 26(3): 689-98. [http://dx.doi.org/10.1002/ajmg.1320260324] [PMID: 3565483] |
| [46] | Anderson RA, Bryson GM, Parks JS. Lysosomal acid lipase mutations that determine phenotype in Wolman and cholesterol ester storage disease. Mol Genet Metab 1999; 68(3): 333-45. [http://dx.doi.org/10.1006/mgme.1999.2904] [PMID: 10562460] |
| [47] | Scott SA, Liu B, Nazarenko I, et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology 2013; 58(3): 958-65. [http://dx.doi.org/10.1002/hep.26327] [PMID: 23424026] |
| [48] | Lohse P, Chahrokh-Zadeh S, Lohse P, Seidel D. Human lysosomal acid lipase/cholesteryl ester hydrolase and human gastric lipase: identification of the catalytically active serine, aspartic acid, and histidine residues. J Lipid Res 1997; 38(5): 892-903. [PMID: 9186907] |
| [49] | Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J Biol Chem 1975; 250(21): 8487-95. [PMID: 172501] |
| [50] | Miller WL, Bose HS. Early steps in steroidogenesis: intracellular cholesterol trafficking. J Lipid Res 2011; 52(12): 2111-35. [http://dx.doi.org/10.1194/jlr.R016675] [PMID: 21976778] |
| [51] | Salvayre R, Negre A, Maret A, Radom J, Douste-Blazy L. Extracellular origin of the lipid lysosomal storage in cultured fibroblasts from Wolmans disease. Eur J Biochem 1987; 170(1-2): 453-8. [http://dx.doi.org/10.1111/j.1432-1033.1987.tb13721.x] [PMID: 3691534] |
| [52] | Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiencyan under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014; 235(1): 21-30. [http://dx.doi.org/10.1016/j.atherosclerosis.2014.04.003] [PMID: 24792990] |
| [53] | Zschenker O, Jung N, Rethmeier J, et al. Characterization of lysosomal acid lipase mutations in the signal peptide and mature polypeptide region causing Wolman disease. J Lipid Res 2001; 42(7): 1033-40. [PMID: 11441129] |
| [54] | Aslanidis C, Ries S, Fehringer P, Büchler C, Klima H, Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics 1996; 33(1): 85-93. [http://dx.doi.org/10.1006/geno.1996.0162] [PMID: 8617513] |
| [55] | Pagani F, Pariyarath R, Garcia R, et al. New lysosomal acid lipase gene mutants explain the phenotype of Wolman disease and cholesteryl ester storage disease. J Lipid Res 1998; 39(7): 1382-8. [PMID: 9684740] |
| [56] | Klima H, Ullrich K, Aslanidis C, Fehringer P, Lackner KJ, Schmitz G. A splice junction mutation causes deletion of a 72-base exon from the mRNA for lysosomal acid lipase in a patient with cholesteryl ester storage disease. J Clin Invest 1993; 92(6): 2713-8. [http://dx.doi.org/10.1172/JCI116888] [PMID: 8254026] |
| [57] | Ameis D, Brockmann G, Knoblich R, et al. A 5 splice-region mutation and a dinucleotide deletion in the lysosomal acid lipase gene in two patients with cholesteryl ester storage disease. J Lipid Res 1995; 36(2): 241-50. [PMID: 7751811] |
| [58] | Roussel A, Canaan S, Egloff M-P, et al. Crystal structure of human gastric lipase and model of lysosomal acid lipase, two lipolytic enzymes of medical interest. J Biol Chem 1999; 274(24): 16995-7002. [http://dx.doi.org/10.1074/jbc.274.24.16995] [PMID: 10358049] |
| [59] | Pagani F, Garcia R, Pariyarath R, et al. Expression of lysosomal acid lipase mutants detected in three patients with cholesteryl ester storage disease. Hum Mol Genet 1996; 5(10): 1611-7. [http://dx.doi.org/10.1093/hmg/5.10.1611] [PMID: 8894696] |
| [60] | Eto Y, Kitagawa T. Wolmans disease with hypolipoproteinemia and acanthocytosis: clinical and biochemical observations. J Pediatr 1970; 77(5): 862-7. [http://dx.doi.org/10.1016/S0022-3476(70)80248-7] [PMID: 5504079] |
| [61] | Rao RH, Mansbach CM II. Acid lipase in rat intestinal mucosa: physiological parameters. Biochim Biophys Acta 1990; 1043(3): 273-80. [http://dx.doi.org/10.1016/0005-2760(90)90027-U] [PMID: 2322572] |
| [62] | Meyers W, Hoeg J, Demosky S, Herbst J, Brewer H. The use of parenteral hyperalimentation and elemental formula feeding in the treatment of wolman disease. Nutr Res 1985; 5: 423-9. [http://dx.doi.org/10.1016/S0271-5317(85)80226-8] |
| [63] | Assmann G, Fredrickson DS, Sloan HR, Fales HM, Highet RJ. Accumulation of oxygenated steryl esters in Wolmans disease. J Lipid Res 1975; 16(1): 28-38. [PMID: 162929] |
| [64] | Fitoussi G, Nègre-Salvayre A, Pieraggi MT, Salvayre R. New pathogenetic hypothesis for Wolman disease: possible role of oxidized low-density lipoproteins in adrenal necrosis and calcification. Biochem J 1994; 301(Pt 1): 267-73. [http://dx.doi.org/10.1042/bj3010267] [PMID: 8037680] |
| [65] | Nègre-Salvayre A, Salvayre R. UV-treated lipoproteins as a model system for the study of the biological effects of lipid peroxides on cultured cells. 4. Calcium is involved in the cytotoxicity of UV-treated LDL on lymphoid cell lines. Biochim Biophys Acta 1992; 1123(2): 207-15. [http://dx.doi.org/10.1016/0005-2760(92)90113-A] [PMID: 1739749] |
| [66] | Marshall WC, Ockenden BG, Fosbrooke AS, Cumings JN. Wolmans disease. A rare lipidosis with adrenal calcification. Arch Dis Child 1969; 44(235): 331-41. [http://dx.doi.org/10.1136/adc.44.235.331] [PMID: 5785183] |
| [67] | Lowden JA, Barson AJ, Wentworth P. Wolmans disease: a microscopic and biochemical study showing accumulation of ceroid and esterified cholesterol. Can Med Assoc J 1970; 102(4): 402-5. [PMID: 5414926] |
| [68] | Lough J, Fawcett J, Wiegensberg B. Wolmans disease. An electron microscopic, histochemical, and biochemical study. Arch Pathol 1970; 89(2): 103-10. [PMID: 5412920] |
| [69] | Byrd JC III, Powers JM. Wolmans disease: ultrastructural evidence of lipid accumulation in central and peripheral nervous systems. Acta Neuropathol 1979; 45(1): 37-42. [http://dx.doi.org/10.1007/BF00691802] [PMID: 216225] |
| [70] | Kamoshita S, Landing BH. Distribution of lesions in myenteric plexus and gastrointestinal mucosa in lipidoses and other neurologic disorders of children. Am J Clin Pathol 1968; 49(3): 312-8. [http://dx.doi.org/10.1093/ajcp/49.3.312] [PMID: 5645093] |
| [71] | Stein J, Garty BZ, Dror Y, Fenig E, Zeigler M, Yaniv I. Successful treatment of Wolman disease by unrelated umbilical cord blood transplantation. Eur J Pediatr 2007; 166(7): 663-6. [http://dx.doi.org/10.1007/s00431-006-0298-6] [PMID: 17033804] |
| [72] | Osellame LD, Duchen MR. Quality control gone wrong: mitochondria, lysosomal storage disorders and neurodegeneration. Br J Pharmacol 2014; 171(8): 1958-72. [http://dx.doi.org/10.1111/bph.12453] [PMID: 24116849] |
| [73] | Guazzi GC, Martin JJ, Philippart M, et al. Wolmans disease. Eur Neurol 1968; 1(6): 334-62. [http://dx.doi.org/10.1159/000113673] [PMID: 5711398] |
| [74] | Gasche C, Aslanidis C, Kain R, et al. A novel variant of lysosomal acid lipase in cholesteryl ester storage disease associated with mild phenotype and improvement on lovastatin. J Hepatol 1997; 27(4): 744-50. [http://dx.doi.org/10.1016/S0168-8278(97)80092-X] [PMID: 9365051] |
| [75] | Bindu PS, Taly AB, Christopher R, et al. Cholesterol ester storage disease with unusual neurological manifestations in two siblings: a report from South India. J Child Neurol 2007; 22(12): 1401-4. [http://dx.doi.org/10.1177/0883073807307104] [PMID: 18174560] |
| [76] | Tolar J, Petryk A, Khan K, et al. Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease. Bone Marrow Transplant 2009; 43(1): 21-7. [http://dx.doi.org/10.1038/bmt.2008.273] [PMID: 18776925] |
| [77] | Krivit W, Peters C, Dusenbery K, et al. Wolman disease successfully treated by bone marrow transplantation. Bone Marrow Transplant 2000; 26(5): 567-70. [http://dx.doi.org/10.1038/sj.bmt.1702557] [PMID: 11019848] |
| [78] | Yanir A, Allatif MA, Weintraub M, Stepensky P. Unfavorable outcome of hematopoietic stem cell transplantation in two siblings with Wolman disease due to graft failure and hepatic complications. Mol Genet Metab 2013; 109(2): 224-6. [http://dx.doi.org/10.1016/j.ymgme.2013.03.007] [PMID: 23583223] |
| [79] | Gramatges MM, Dvorak CC, Regula DP, Enns GM, Weinberg K, Agarwal R. Pathological evidence of Wolmans disease following hematopoietic stem cell transplantation despite correction of lysosomal acid lipase activity. Bone Marrow Transplant 2009; 44(7): 449-50. [http://dx.doi.org/10.1038/bmt.2009.57] [PMID: 19308038] |
| [80] | Muretto P, Del Fiasco S, Angelucci E, De Rosa F, Lucarelli G. Bone marrow transplantation in thalassemia: modifications of hepatic iron overload and associated lesions after long-term engrafting. Liver 1994; 14(1): 14-24. [http://dx.doi.org/10.1111/j.1600-0676.1994.tb00002.x] [PMID: 8177025] |
| [81] | Krivit W, Freese D, Chan KW, Kulkarni R. Wolmans disease: a review of treatment with bone marrow transplantation and considerations for the future. Bone Marrow Transplant 1992; 10(Suppl. 1): 97-101. [PMID: 1521099] |
| [82] | Arterburn JN, Lee WM, Wood RP, Shaw BW, Markin RS. Orthotopic liver transplantation for cholesteryl ester storage disease. J Clin Gastroenterol 1991; 13(4): 482-5. [http://dx.doi.org/10.1097/00004836-199108000-00028] [PMID: 1918862] |
| [83] | Ferry GD, Whisennand HH, Finegold MJ, Alpert E, Glombicki A. Liver transplantation for cholesteryl ester storage disease. J Pediatr Gastroenterol Nutr 1991; 12(3): 376-8. [http://dx.doi.org/10.1097/00005176-199104000-00016] [PMID: 2072231] |
| [84] | Kale AS, Ferry GD, Hawkins EP. End-stage renal disease in a patient with cholesteryl ester storage disease following successful liver transplantation and cyclosporine immunosuppression. J Pediatr Gastroenterol Nutr 1995; 20(1): 95-7. [http://dx.doi.org/10.1097/00005176-199501000-00016] [PMID: 7884624] |
| [85] | Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiencymacrophage-targeted glucocerebrosidase for Gauchers disease. N Engl J Med 1991; 324(21): 1464-70. [http://dx.doi.org/10.1056/NEJM199105233242104] [PMID: 2023606] |
| [86] | Braulke T, Bonifacino J. Sorting of lysosomal proteins. Biochim et Biophys Acta 2009; 1793(4): 605-14. [http://dx.doi.org/10.1016/j.bbamcr.2008.10.016] |
| [87] | Lachmann RH. Enzyme replacement therapy for lysosomal storage diseases. Curr Opin Pediatr 2011; 23(6): 588-93. [http://dx.doi.org/10.1097/MOP.0b013e32834c20d9] [PMID: 21946346] |
| [88] | Poznansky MJ, Hutchison SK, Davis PJ. Enzyme replacement therapy in fibroblasts from a patient with cholesteryl ester storage disease. FASEB J 1989; 3(2): 152-6. [PMID: 2644147] |
| [89] | Sando GN, Henke VL. Recognition and receptor-mediated endocytosis of the lysosomal acid lipase secreted by cultured human fibroblasts. J Lipid Res 1982; 23(1): 114-23. [PMID: 7057100] |
| [90] | Mistry PK, Wraight EP, Cox TM. Therapeutic delivery of proteins to macrophages: implications for treatment of Gauchers disease. Lancet 1996; 348(9041): 1555-9. [http://dx.doi.org/10.1016/S0140-6736(96)04451-0] [PMID: 8950883] |
| [91] | FDA approves first drug to treat a rare enzyme disorder in pediatric and adult patients. US Department of Health and Human Services: USA. 2015. |
| [92] | Burton BK, Balwani M, Feillet F, et al. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med 2015; 373(11): 1010-20. [http://dx.doi.org/10.1056/NEJMoa1501365] [PMID: 26352813] |
| [93] | Valyannopoulos V, Plantaz D, Vara R, et al. Clinical effect of sebelipase alfa on survival and growth in infants with lysosomal acid lipase deficiency (Wolman disease). Mol Genet Metab 2014; 111: S108. [http://dx.doi.org/10.1016/j.ymgme.2013.12.268] |
| [94] | Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation 2000; 101(2): 207-13. [http://dx.doi.org/10.1161/01.CIR.101.2.207] [PMID: 10637210] |
| [95] | Ginsberg HN, Le NA, Short MP, Ramakrishnan R, Desnick RJ. Suppression of apolipoprotein B production during treatment of cholesteryl ester storage disease with lovastatin. Implications for regulation of apolipoprotein B synthesis. J Clin Invest 1987; 80(6): 1692-7. [http://dx.doi.org/10.1172/JCI113259] [PMID: 3680522] |
| [96] | Dalgiç B, Sari S, Gündüz M, et al. Cholesteryl ester storage disease in a young child presenting as isolated hepatomegaly treated with simvastatin. Turk J Pediatr 2006; 48(2): 148-51. [PMID: 16848116] |
| [97] | Tarantino MD, McNamara DJ, Granstrom P, Ellefson RD, Unger EC, Udall JN Jr. Lovastatin therapy for cholesterol ester storage disease in two sisters. J Pediatr 1991; 118(1): 131-5. [http://dx.doi.org/10.1016/S0022-3476(05)81866-9] [PMID: 1986080] |
| [98] | Glueck CJ, Lichtenstein P, Tracy T, Speirs J. Safety and efficacy of treatment of pediatric cholesteryl ester storage disease with lovastatin. Pediatr Res 1992; 32(5): 559-65. [http://dx.doi.org/10.1203/00006450-199211000-00015] [PMID: 1480458] |
| [99] | Di Bisceglie AM, Ishak KG, Rabin L, Hoeg JM. Cholesteryl ester storage disease: hepatopathology and effects of therapy with lovastatin. Hepatology 1990; 11(5): 764-72. [http://dx.doi.org/10.1002/hep.1840110509] [PMID: 2347551] |
| [100] | Tadiboyina VT, Liu DM, Miskie BA, Wang J, Hegele RA. Treatment of dyslipidemia with lovastatin and ezetimibe in an adolescent with cholesterol ester storage disease. Lipids Health Dis 2005; 4: 26-32. [http://dx.doi.org/10.1186/1476-511X-4-26] [PMID: 16255772] |
| [101] | Yokoyama S, McCoy E. Long-term treatment of a homozygous cholesteryl ester storage disease with combined cholestyramine and lovastatin. J Inherit Metab Dis 1992; 15(2): 291-2. [http://dx.doi.org/10.1007/BF01799650] [PMID: 1528002] |
| [102] | Levy R, Ostlund RE Jr, Schonfeld G, Wong P, Semenkovich CF. Cholesteryl ester storage disease: complex molecular effects of chronic lovastatin therapy. J Lipid Res 1992; 33(7): 1005-15. [PMID: 1431581] |
| [103] | Iverson SA, Cairns SR, Ward CP, Fensom AH. Asymptomatic cholesteryl ester storage disease in an adult controlled with simvastatin. Ann Clin Biochem 1997; 34(Pt 4): 433-6. [http://dx.doi.org/10.1177/000456329703400418] [PMID: 9247680] |
| [104] | Macarron R, Banks MN, Bojanic D, et al. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov 2011; 10(3): 188-95. [http://dx.doi.org/10.1038/nrd3368] [PMID: 21358738] |
| [105] | Huang H-P, Chuang C-Y, Kuo H-C. Induced pluripotent stem cell technology for disease modeling and drug screening with emphasis on lysosomal storage diseases. Stem Cell Res Ther 2012; 3(4): 34. [http://dx.doi.org/10.1186/scrt125] [PMID: 22925465] |
| [106] | Xu M, Liu K, Swaroop M, et al. A phenotypic compound screening assay for lysosomal storage diseases. J Biomol Screen 2014; 19(1): 168-75. [http://dx.doi.org/10.1177/1087057113501197] [PMID: 23983233] |
| [107] | Xu M, Liu K, Swaroop M, et al. δ-Tocopherol reduces lipid accumulation in Niemann-Pick type C1 and Wolman cholesterol storage disorders. J Biol Chem 2012; 287(47): 39349-60. [http://dx.doi.org/10.1074/jbc.M112.357707] [PMID: 23035117] |
| [108] | Du H, Duanmu M, Witte D, Grabowski GA. Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Hum Mol Genet 1998; 7(9): 1347-54. [http://dx.doi.org/10.1093/hmg/7.9.1347] [PMID: 9700186] |
| [109] | Lemonnier T, Blanchard S, Toli D, et al. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum Mol Genet 2011; 20(18): 3653-66. [http://dx.doi.org/10.1093/hmg/ddr285] [PMID: 21685203] |
| [110] | Zhang J, Lian Q, Zhu G, et al. A human iPSC model of Hutchinson Gilford Progeria reveals vascular smooth muscle and mesenchymal stem cell defects. Cell Stem Cell 2011; 8(1): 31-45. [http://dx.doi.org/10.1016/j.stem.2010.12.002] [PMID: 21185252] |
| [111] | Yu D, Swaroop M, Wang M, et al. Niemann-pick disease type C: induced pluripotent stem cell-derived neuronal cells for modeling neural disease and evaluating drug efficacy. J Biomol Screen 2014; 19(8): 1164-73. [http://dx.doi.org/10.1177/1087057114537378] [PMID: 24907126] |
| [112] | Kuriyama M, Yoshida H, Suzuki M, Fujiyama J, Igata A. Lysosomal acid lipase deficiency in rats: lipid analyses and lipase activities in liver and spleen. J Lipid Res 1990; 31(9): 1605-12. [PMID: 2246613] |
| [113] | Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126(4): 663-76. [http://dx.doi.org/10.1016/j.cell.2006.07.024] [PMID: 16904174] |
| [114] | Dimos JT, Rodolfa KT, Niakan KK, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008; 321(5893): 1218-21. [http://dx.doi.org/10.1126/science.1158799] [PMID: 18669821] |
| [115] | Spence J, Mayhew C, Rankin S, et al. Directed differentiation of human pluripotent stem cells into intestinal tissues in vitro. Nature 2011; 470(7332): 105-9. [http://dx.doi.org/10.1038/nature09691] |
| [116] | Chen Y-F, Tseng C-Y, Wang H-W, Kuo H-C, Yang VW, Lee OK. Rapid generation of mature hepatocyte-like cells from human induced pluripotent stem cells by an efficient three-step protocol. Hepatology 2012; 55(4): 1193-203. [http://dx.doi.org/10.1002/hep.24790] [PMID: 22095466] |
| [117] | Meng X-L, Shen J-S, Kawagoe S, Ohashi T, Brady RO, Eto Y. Induced pluripotent stem cells derived from mouse models of lysosomal storage disorders. Proc Natl Acad Sci USA 2010; 107(17): 7886-91. [http://dx.doi.org/10.1073/pnas.1002758107] [PMID: 20385825] |
| [118] | Huang H-P, Chen P-H, Hwu W-L, et al. Human Pompe disease-induced pluripotent stem cells for pathogenesis modeling, drug testing and disease marker identification. Hum Mol Genet 2011; 20(24): 4851-64. [http://dx.doi.org/10.1093/hmg/ddr424] [PMID: 21926084] |
| [119] | Maetzel D, Sarkar S, Wang H, et al. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Rep 2014; 2(6): 866-80. [http://dx.doi.org/10.1016/j.stemcr.2014.03.014] [PMID: 24936472] |
| [120] | Swaroop M, Thorne N, Rao MS, Austin CP, McKew JC, Zheng W. Evaluation of cholesterol reduction activity of methyl-β-cyclodextrin using differentiated human neurons and astrocytes. J Biomol Screen 2012; 17(9): 1243-51. [http://dx.doi.org/10.1177/1087057112456877] [PMID: 22923786] |
| [121] | Smid BE, Aerts JM, Boot RG, Linthorst GE, Hollak CE. Pharmacological small molecules for the treatment of lysosomal storage disorders. Expert Opin Investig Drugs 2010; 19(11): 1367-79. [http://dx.doi.org/10.1517/13543784.2010.524205] [PMID: 20942596] |
| [122] | Mistry PK, Lukina E, Ben Turkia H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA 2015; 313(7): 695-706. [http://dx.doi.org/10.1001/jama.2015.459] [PMID: 25688781] |
| [123] | Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 2003; 4(3): 181-91. [http://dx.doi.org/10.1038/nrm1052] [PMID: 12612637] |
| [124] | Valenzano KJ, Khanna R, Powe AC, et al. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay Drug Dev Technol 2011; 9(3): 213-35. [http://dx.doi.org/10.1089/adt.2011.0370] [PMID: 21612550] |
| [125] | Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2011; 8(2): 108-17. [http://dx.doi.org/10.1038/nrneurol.2011.200] [PMID: 22187000] |
| [126] | Appelqvist H, Wäster P, Kågedal K, Öllinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol 2013; 5(4): 214-26. [http://dx.doi.org/10.1093/jmcb/mjt022] [PMID: 23918283] |
| [127] | Wraith JE. Limitations of enzyme replacement therapy: current and future. J Inherit Metab Dis 2006; 29(2-3): 442-7. [http://dx.doi.org/10.1007/s10545-006-0239-6] [PMID: 16763916] |
| [128] | Lohse P, Maas S, Sewell AC, Seidel D, van Diggelen O, Seidel D. Molecular defects underlying Wolman disease appear to be more heterogeneous than those resulting in cholesteryl ester storage disease. J Lipid Res 1999; 40(2): 221-8. [PMID: 9925650] |
| [129] | Fujiyama J, Sakuraba H, Kuriyama M, et al. A new mutation (LIPA Tyr22X) of lysosomal acid lipase gene in a Japanese patient with Wolman disease. Hum Mutat 1996; 8(4): 377-80. [http://dx.doi.org/10.1002/(SICI)1098-1004(1996)8:4<377::AID-HUMU15>3.0.CO;2-#] [PMID: 8956047] |
| [130] | Anderson RA, Byrum RS, Coates PM, Sando GN. Mutations at the lysosomal acid cholesteryl ester hydrolase gene locus in Wolman disease. Proc Natl Acad Sci USA 1994; 91(7): 2718-22. [http://dx.doi.org/10.1073/pnas.91.7.2718] [PMID: 8146180] |
| [131] | Ries S, Aslanidis C, Fehringer P, Carel JC, Gendrel D, Schmitz G. A new mutation in the gene for lysosomal acid lipase leads to Wolman disease in an African kindred. J Lipid Res 1996; 37(8): 1761-5. [PMID: 8864960] |
| [132] | Lin P, Raikar S, Jimenez J, Conard K, Furuya K. Novel mutation in a patient with cholesterol ester storage disease. Case Rep Genet 2015. Epub ahead of print. [http://dx.doi.org/10.1155/2015/347342] |
| [133] | Hooper AJ, Tran HA, Formby MR, Burnett JR. A novel missense LIPA gene mutation, N98S, in a patient with cholesteryl ester storage disease. Clin Chim Acta 2008; 398(1-2): 152-4. [http://dx.doi.org/10.1016/j.cca.2008.08.007] [PMID: 18775687] |
| [134] | Ries S, Büchler C, Schindler G, et al. Different missense mutations in histidine-108 of lysosomal acid lipase cause cholesteryl ester storage disease in unrelated compound heterozygous and hemizygous individuals. Hum Mutat 1998; 12(1): 44-51. [http://dx.doi.org/10.1002/(SICI)1098-1004(1998)12:1<44::AID-HUMU7>3.0.CO;2-O] [PMID: 9633819] |
| [135] | Kojima S, Watanabe N, Takashimizu S, et al. Senescent case of cholesterol ester storage disease that progressed to liver cirrhosis with a novel mutation (N250H) of lysosomal acid lipase gene. Hepatol Res 2013; 43(12): 1361-7. [http://dx.doi.org/10.1111/hepr.12087] [PMID: 23675960] |
| [136] | Kuranobu N, Murakami J, Okamoto K, et al. Cholesterol ester storage disease with a novel LIPA mutation (L264P) that presented massive hepatomegaly: a case report. Hepatol Res 2015. [PMID: 26385844] |
| [137] | Redonnet-Vernhet I, Chatelut M, Basile J-P, Salvayre R, Levade T. Cholesteryl ester storage disease: relationship between molecular defects and in situ activity of lysosomal acid lipase. Biochem Mol Med 1997; 62(1): 42-9. [http://dx.doi.org/10.1006/bmme.1997.2626] [PMID: 9367797] |
| [138] | Lohse P, Maas S, Lohse P, et al. Compound heterozygosity for a Wolman mutation is frequent among patients with cholesteryl ester storage disease. J Lipid Res 2000; 41(1): 23-31. [PMID: 10627498] |
| [139] | Seedorf U, Wiebusch H, Muntoni S, et al. A novel variant of lysosomal acid lipase (Leu336>Pro) associated with acid lipase deficiency and cholesterol ester storage disease. Arterioscler Thromb Vasc Biol 1995; 15(6): 773-8. [http://dx.doi.org/10.1161/01.ATV.15.6.773] [PMID: 7773732] |