- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Utilizing HaloTag Technology to Track the Fate of PCSK9 from Intracellular vs. Extracellular Sources
Xi Ai1, *, Paul Fischer2, Oksana C Palyha1, Douglas Wisniewski2, Brian Hubbard1, Karen Akinsanya1, Alison M Strack1, Anka G Ehrhardt2, *
Abstract
The function of a particular protein is dependent upon its localization and milieu. The ability to track the "fate" of a protein is a valuable tool to elucidate its function. We present the use of HaloTag technology to study the localization and fate of human Proprotein Convertase Subtilisin-like Kexin type 9 (PCSK9).
The role of PCSK9 in the regulation of circulating low density lipoprotein-cholesterol (LDL-c) levels is ascribed to binding of circulating PCSK9 to the LDL receptor (LDLR) and subsequent lysosomal degradation of LDLR. However, hints in the literature indicate that intracellular PCSK9 may act on the LDLR, possibly during processing of newly synthesized protein. To address this question, the source and fate of intracellular PCSK9 requires further investigation.
We applied HaloTag technology to distinguish the source of intracellular PCSK9 and showed that newly synthesized intracellular PCSK9 has unique localization from the PCSK9 after re-uptake. This suggests different functions of PCSK9 while interacting with the LDLR.
Article Information
Identifiers and Pagination:
Year: 2012Volume: 6
Issue: Suppl 1
First Page: 38
Last Page: 47
Publisher Id: CCGTM-6-38
DOI: 10.2174/1875397301206010038
Article History:
Received Date: 13/4/2012Revision Received Date: 16/5/2012
Acceptance Date: 16/5/2012
Electronic publication date: 20/9/2012
Collection year: 2012
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to these authors at the Department of Atherosclerosis, RY80T-A100, Merck Research Laboratories, 126 E. Lincoln Ave, Rahway, NJ 07065, USA; Tel: 732-594-2286; Fax: 732-594-2510; E-mail: xi.ai@merck.com; Department of In Vitro Pharmacology, RY80N-68A, Merck Research Laboratories, 126 E. Lincoln Ave, Rahway, NJ 07065, USA; Tel: 732- 594-5530; Fax: 908-823-3127; E-mail: anka.ehrhardt@merck.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 13-4-2012 |
Original Manuscript | Utilizing HaloTag Technology to Track the Fate of PCSK9 from Intracellular vs. Extracellular Sources | |
INTRODUCTION
Cellular function is tightly regulated by spatio-temporal organization. Cell organelles can be seen as units within this organization. They maintain spaces that allow distinct environmental conditions enabling specific proteins to function only at the time they exist within a particular organelle. Lysosomes, for example, are organelles that create an acidic environment, allowing for the regulated function of proteases and degradation of cellular components [1Alberts B, Johnson A, Lewis J, et al. Transport from the Trans Golgi network to lysosomes In: molecular biology of the cell. 4th. New York : Garland Science 2012.]. Proteins are regulated by their fate, i.e. the order in which they pass through various organelles. In these cases, some post-translational modifications are dependent on a protein passing from the endoplasmic reticulum (ER) to the Golgi apparatus. Understanding the function of a protein in the cell is often tied to the understanding of its current localization, the protein's origin and fate of a protein [1Alberts B, Johnson A, Lewis J, et al. Transport from the Trans Golgi network to lysosomes In: molecular biology of the cell. 4th. New York : Garland Science 2012.].
Studying the destiny of a cellular protein is usually hindered by the presence of molecules of the same protein in multiple places at the same time, with the previous position of any individual molecule unknown. In order to elucidate the fate of small groups of molecules, multiple methods for protein tracking are being employed in modern cell biology. These techniques include fluorescent protein labels [2Cubitt AB, Heim R, Adams SR, Boyd AE, Gross LA, Tsien RY. Understanding, improving and using green fluorescent proteins Trends Biochem Sci 1995; 20: 448-55.], fluorescence recovery after photo-bleaching (FRAP) [3Walczak CE, Rizk RS, Shaw SL. The use of fluorescence redistribution after photobleaching for analysis of cellular microtubule dynamics Methods Cell Biol 2010; 97: 35-52.], photoactivation [4Lohmann D, Petrak K. Photoactivation and photocontrolled release of bioactive materials Crit Rev Ther Drug Carrier Syst 1989; 5: 263-320.], surface biotinylation [5Arancibia-Cárcamo IL, Fairfax BP, Moss SJ, Kittler JT. Studying the Localization, surface stability and endocytosis of neurotransmitter receptors by antibody labeling and biotinylation approaches In: Kittler JT, Moss SJ, Eds. The dynamic synapse: molecular methods in ionotropic receptor biology. Boca Raton (FL) : CRC Press 2012.] and photo-switchable proteins [6Henderson JN, Remington SJ. The kindling fluorescent protein: a transient photoswitchable marker Physiology 2006; 21: 162-70.]. These types of studies require sophisticated instrumentation and represent significant technical challenges for the modern cell biologist.
Over the last few years, technologies employing genetically encoded protein tags, for which specific binding ligands exist, have opened new possibilities in cell biology [7Johnsson N, Johnsson K. A fusion of disciplines: Chemical approaches to exploit fusion proteins for functional genomics Chembiochem 2003; 4: 803-10.]. One example of such technology is a commercially available HaloTag system [8Los GV, Zimprich C, McDougall MG, et al. The HaloTag (TM): a novel technology for cellular analysis J Neurochem 2005; 94: 15.]. This 33 kDa protein is engineered to covalently and irreversibly bind specific ligands. In combination with a collection of ligands displaying a range of different properties, the HaloTag technology allows new tracking studies with relatively simple tools [8Los GV, Zimprich C, McDougall MG, et al. The HaloTag (TM): a novel technology for cellular analysis J Neurochem 2005; 94: 15., 9Los GV, Encell LP, McDougall MG, et al. HatoTag: a novel protein labeling technology for cell imaging and protein analysis ACS Chem Biol 2008; 3: 373-82.].
In this study, we utilize two main features of the available HaloTag ligands, their spectral differences and their different cell permeability, to study the fate of Proprotein Convertase Subtilisin-like Kexin type 9 (PCSK9). PCSK9 has become an attractive target for treatment of dyslipidemia [10Hedrick JA. Targeting PCSK9 for the treatment of hypercholesterolemia Curr Opin Investig Drugs 2009; 10: 938-46.-12Seidah NG. PCSK9 as a therapeutic target of dyslipidemia Expert Opin Ther Targets 2009; 13: 19-28.]. Human genetic data for PCSK9 have provided strong validation for lowering low density lipoprotein-cholesterol (LDL-c) and improved coronary heart disease °CHD) mortality. Multiple studies, including the Dallas Heart Study, have shown that the gain of function mutations of PCSK9 have increased plasma LDL-c levels and early onset of myocardial infarction (MI), whereas the loss of function mutations of PCSK9 have decreased plasma LDL-c levels and reduced incidence of CHD [13Abifadel M, Varret M, Rabes JP A, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia Nat Genet 2003; 34: 154-6.-15Victor RG, Haley RW, Willett DL, et al. The dallas heart study: a population-based probability sample for the multidisciplinary study of ethnic differences in cardiovascular health Am J Cardiol 2004; 93: 1473-80.].
The mechanism of PCSK9 function has been extensively explored [11Horton JD, Cohen JC, Hobbs HH. PCSK9: a convertase that coordinates LDL catabolism J Lipid Res 2009; 50: S172-7.,16Lagace TA, Curtis DE, Garuti R, et al. Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice J Clin Investig 2006; 116: 2995-3005.-18Maxwell KN, Breslow JL. Proprotein convertase subtilisin kexin 9: the third locus implicated in autosomal dominant hypercholesterolemia Curr Opin Lipidol 2005; 16: 167-72.]. Immediately after being synthesized, PCSK9 undergoes autocatalytic cleavage, through which a 10 kDa prodomain is cleaved but remains non-covalently bound to the 63 kDa mature protein. Once secreted, the cleaved PCSK9 binds to low density lipoprotein receptors (LDLR), and subsequently directs the LDLR to lysosomes for degradation in the liver, thus controlling the levels of LDL-c in plasma. In addition to the well established secretion pathway, PCSK9 has also been implicated in regulating LDLR through an intracellular pathway in which intracellular PCSK9 binds to LDLR prior to secretion thus enhancing LDLR degradation [19Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment Proc Natl Acad Sci USA 2005; 102: 2069-74.-21Poirier S, Mayer G, Poupon V, et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation evidence for an intracellular route J Biol Chem 2009; 284: 28856-64.]. To shed light on the PCSK9 function in both pathways, we utilized HaloTag (HT) technology recently developed by Promega [8Los GV, Zimprich C, McDougall MG, et al. The HaloTag (TM): a novel technology for cellular analysis J Neurochem 2005; 94: 15.] to study PCSK9 trafficking in a hepatoma cell line. In the current study, we established a Huh7 stable cell line expressing C-terminal PCSK9-HaloTag (PCSK9-HT) fusion protein. Through labeling cells with either cell impermeable (Alexa Fluor 488) or permeable (TMR) fluorescent ligands, we were able to distinguish the pool of newly synthesized intracellular PCSK9 from that of secreted and then internalized PCSK9. We also investigated the interaction of both intracellular and internalized PCSK9 with LDLR, as well as their sub-cellular localization. In this study, the HaloTag technology has enabled us to distinguish the source and fate of two pools of PCSK9 in living cells. The model system established here allows us to further understand and elucidate the mechanism of PCSK9 function.
MATERIALS AND METHODOLOGY
Establishment of stable cell line
The plasmid pFC14A-PCSK9 was produced in three steps. First, SgfI and PmeI endonuclease sites were introduced into the 5'-end or 3'-end of the human PCSK9 gene, respectively, by PCR using the oligonucleotides 5'- CA-CGGCGATCGCCATGGGCACCGTCAGCTCCAGGC-3' (5' primer) and 5'-CTTGGTTTAAACCTGGAGCTCCT-GGGAGGCCTGC-3' (3' primer), and pCDNA3.1/V5-His-PCSK9 [22Fisher TS, Lo Surdo P, Pandit S, et al. Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation J Biol Chem 2007; 282: 20502-12.] as a template. Next, the amplified PCSK9 ORF was sequenced and cloned into the pF4K CMV Flexi Vector (Promega, WI, USA) following the standard cloning procedure [23Russell DW, Sambrook J. Molecular cloning: a laboratory manual 2001.]. The resulting plasmid was sequenced and cloned into the pFC14A HaloTag CMV Flexi Vector (Promega) to produce pFC14A-PCSK9. Prior to transfection, pFC14A-PCSK9 and pCDNA3.1/hygro/lacZ (Invitrogen, NY, USA) were linearized with BglII (New England Biolabs, MA, USA). Huh7 cells were seeded on day 0 at 1.5×106 cells in 60 mm dishes and cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS, v/v, Invitrogen) at 37°C and 5% CO2 overnight. On day 1, the cells were co-transfected with linearized pFC14A-PCSK9 and pCDNA3.1/hygro/lacZ using GenJet In vitro DNA transfection reagent for Huh7 cells (Ver. II) (SignaGen Laboratories, MD, USA) following the manufacturer's instructions. At 48 h post transfection (day 3), the cells were fed with selective media (DMEM supplemented with 10% FBS and 200 µg/ml Hygromycin B [Invitrogen]), and re-plated onto 100 mm dishes. On day 17, cells growing in selective media were collected and labeled with HaloTag R110Direct Ligand (Promega) according to the manufacturer's instructions. The cells expressing HaloTag were sorted using Fluorescence-Activated Cell Sorting (FACS) and 8×104 cells were seeded into 6-well culture plates containing selective media. Cells were allowed to grow and expanded at 37°C and 5% CO2 for two weeks. On day 31, cells underwent a second round of FACS. The cells were harvested and labeled as described above. Fluorescence positive cells were seeded into 96-well culture plates at 1 cell/well, and cultured in selective media at 37°C and 5% CO2 until colonies formed. A screen for positive clones was performed as follows: 50 µl of media was transferred from wells containing cell clones to a 96-well assay plate. 1 µl of 0.1 mM HaloTag TMR Ligand (Promega) was added to each well. The plate was incubated at room temperature for 15 min in the dark before adding 10 µl of 6xSDS reducing sample buffer (Boston BioProducts, MA, USA) to each well. Next, 10 µl each of denatured media was loaded onto a 4-12% Bis-Tris SDS-PAGE gel (Invitrogen) and run at 200V for 60 min. The gel was rinsed briefly using Milli-Q H2O followed by image scanning using a Typhoon (GE Healthcare, NJ, USA) at TMR/Cy3 fluorescence setting. Clones showing the PCSK9-HaloTag band on the gel were selected and expanded. To mimic the physiological PCSK9 level, a stable cell line with medium-low PCSK9-HaloTag expression level, known as 14-13-E4, was established. All the experiments described herein were performed using 14-13-E4.
Antibodies
All antibodies were purchased from external vendors: Goat polyclonal antibody against PCSK9 (Abcam, MA, USA), goat polyclonal antibody against LDL receptor (R&D Systems Inc., MN, USA), mouse monoclonal antibody against PDI (Abcam), mouse monoclonal antibody against Golgin97 (Invitrogen), rabbit polyclonal antibody against EEA1 (Abcam), and mouse monoclonal antibody against LAMP1 (Abcam). The following antibodies were used as isotype controls: mouse IgG1 (BD Biosciences, CA, USA), rabbit IgG (Invitrogen) and goat IgG (Invitrogen). Secondary antibodies conjugated with Alexa Fluor were purchased from Invitrogen including: Alexa Fluor 647 donkey-anti-rabbit IgG, Alexa Fluor 647 donkey-anti-mouse IgG, Alexa Fluor 647 donkey-anti-goat IgG, Alexa Fluor 488 donkey-anti-goat IgG, Alexa Fluor 568 donkey-anti-mouse IgG, and Alexa Fluor 568 donkey-anti-rabbit IgG.
HaloTag ligand labeling, Immunofluorescence Analysis and Cell Imaging
For HaloTag ligand labeling, the HaloTag TMR Ligand and Alexa Fluor 488 Ligand (Promega) were used to label the cells expressing PCSK9-HT following the manufacturer's instructions with some modifications. In brief, clone 14-13-E4 or wild type Huh7 cells were plated on 96-well glass bottom culture plates (Matrical Bioscience, WA, USA) and incubated overnight at 37°C and 5% CO2 prior to labeling, with the exception of the experiments where overnight labeling of Alexa Fluor 488 was required. In those experiments, 0.2 µM of Alexa Fluor 488 Ligand was added at the time of cell plating. In the other experiments, 0.2 µM of Alexa Fluor 488 Ligand or 5 µM of TMR ligand was incubated with cells in the DMEM media supplemented with 10% FBS on the following day. The incubation time ranged from 30 to 300 min as required in different experiments for Alexa Fluor 488 and 15 min for TMR. The unbound ligand was washed off with DMEM after labeling. For live cell imaging, the media was replaced with phenol-red free DMEM prior to capturing images. For fixed cell imaging, the labeled cells were fixed with 4% para-formaldehyde containing 0.2 M sucrose in PBS for 10 min at room temperature followed by permeabilization with 0.1% Triton X-100 (in PBS, Boston BioProducts). The cells were then washed in DPBS (no Ca2+ or Mg2+, Invitrogen) three times and incubated overnight with 2% Normal Donkey Serum (Jackson ImmunoResearch Laboratories, PA, USA) and 0.01% Triton X-100 in PBS at 4°C. The cells were incubated for 2 h at room temperature with primary antibodies and then washed three times with DPBS. The corresponding species-specific Alexa Fluor (488, 568, or 647) conjugated secondary antibodies were used to reveal antigen-antibody complexes by incubation for 45 min at room temperature. After three washes in DPBS, the cell nuclei were counterstained with Hoechst 33342 (Invitrogen). Fluorescence was visualized using either a Zeiss LSM 510 confocal microscope equipped with multiple lasers or an Evotech Opera Confocal High Content Imager. Potentially overlapping emissions were collected separately using sequential scanning. On the Zeiss LSM 510, 4 color images (Hoechst, Alexa Fluor 488, Alexa Fluor 568 or TMR, Alexa Fluor 647) were acquired in a two pass configuration. The configuration of pass 1 was as follows: simultaneous 2P (780 nM)/543 laser excitation and 430BP/585BP emission filters plus DIC for transmitted light. The configuration of pass 2 was: 488/633 laser excitation and 515BP/670LP emission filters. Three color images (Alexa Fluor 488, TMR, DRAQ5) were acquired on the Opera as 2 pass images using the following configuration: 488/633 laser excitation 535BP/690BP emission then 568 laser excitation 600BP emission. We also obtained four color images (Hoechst, Alexa Fluor 488, Alexa Fluor 568 or TMR, Alexa Fluor 647) acquired as 2 pass images with the following configuration: 488/633 excitation 535BP/690BP emission, then UV (ARC lamp)/568 laser excitation 600BP emission. All time-lapse and kinetic studies were performed on the Opera using Perkin-Elmer's RCI software.
Purification of HaloTag Fusion PCSK9 (PCSK9-HT)
In the initial step of purification of PCSK9-HT, 800 mL of 14-13-E4 media (lacking phenol red and containing 5% FBS) was applied to a 6 mL Resource Q column (GE Healthcare). The column containing media was washed with seven column volumes of buffer A (25 mM HEPES pH 7.9, 30 mM Sodium Chloride, 0.1 mM Calcium Chloride, 5.0% Glycerol). A salt gradient towards a final of 50% Buffer B (25 mM HEPES pH 7.9, 1.0 M Sodium Chloride, 0.1 mM Calcium Chloride, 5.0% Glycerol) over 20 column volumes was applied for elution. Samples from the peak were run onto 10-20% Tris-Glycine gels and probed with goat polyclonal antibody against PCSK9 (Abcam). The western blot bands were detected with IRDye 800CW donkey-anti-goat IgG (Licor, NE, USA) and scanned with a Licor Odyssey. Positive samples containing PCSK9-HT (~ 270 mM – 350 mM Sodium Chloride) were pooled and concentrated. The concentrated sample was applied to a Superdex 200 10/300GL column (GE Healthcare) and run in buffer containing 25 mM HEPES pH 8.0, 150 mM Sodium Chloride, 0.1 mM Calcium Chloride and 10% glycerol. Samples from the size exclusion chromatography peak were verified by western blot, as noted above, and with Gelcode Blue (Thermo Fisher Scientific, IL, USA) staining. The samples were quantitated using a NanoDrop spectrophotometer and subjected to a LDL uptake assay.
LDL Uptake Assay
The assay was performed as previously described [22Fisher TS, Lo Surdo P, Pandit S, et al. Effects of pH and low density lipoprotein (LDL) on PCSK9-dependent LDL receptor regulation J Biol Chem 2007; 282: 20502-12.] with some modifications. Parental HepG2 cells were seeded into poly-D-lysine coated 96-well plates Corning, NY, USA) at 30, 000 cells/well in DMEM supplemented with 10% FBS (HyClone, UT, USA) in 100 µl volume. After 24 h of incubation at 37°C and 5% CO2, the medium was replaced with 100 µl of DMEM lacking serum and the cells were incubated for additional 24 h. On day 3, the medium was aspirated and 100 µl of Assay Mixture [DMEM + 10% LPDS (Intracel, MD, USA) + 10 µg/ml Alexa Fluor-546 (Invitrogen) labeled human LDL (Intracel) + purified PCSK9 in various concentrations] was applied to the cells. The plates were incubated at 37°C and 5% CO2 for 5 h before being washed with 1xTBS (Bio-Rad, CA, USA) and lysed with 100 µl RIPA buffer (TekNova, CA, USA). The lysate was then transferred to a MicroFluor2 Black “U” bottom plate (Thermo Fisher Scientific). The fluorescent LDL signal was measured using an excitation wavelength of 520 nm and an emission wavelength of 580 nm.
RESULTS
A Stable Cell Line Expressing HaloTag Fusion PCSK9 (PCSK9-HT) was Established
The western blots from cell lysates indicated that the expression level of PCSK9-HT is close to the endogenous level of PCSK9 (~ 2 fold) (Fig. 1A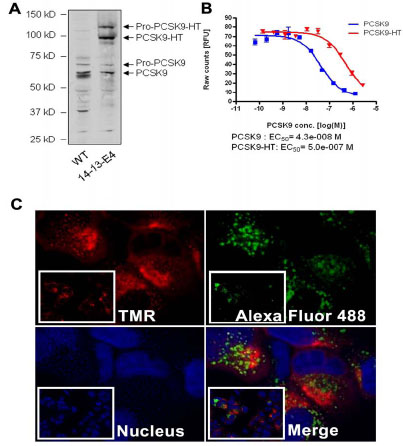 ). To investigate whether introduction of the HaloTag to the C-terminal end of PCSK9 would have an impact on PCSK9 function, PCSK9-HT protein was purified from the culture media of 14-13-E4. The partially purified PCSK9-HT was subjected to the LDL uptake assay. As shown in Fig. (1B), the PCSK9-HT effectively inhibited the LDL receptor mediated LDL uptake in HepG2 cells, confirming that the fusion of the HaloTag to the C-terminal of PCSK9 did not affect the function of PCSK9. The potency of PCSK9-HT was lower when compared to the non-tagged wild type recombinant PCSK9. This effect may be the results of the protein's partial purity.
). To investigate whether introduction of the HaloTag to the C-terminal end of PCSK9 would have an impact on PCSK9 function, PCSK9-HT protein was purified from the culture media of 14-13-E4. The partially purified PCSK9-HT was subjected to the LDL uptake assay. As shown in Fig. (1B), the PCSK9-HT effectively inhibited the LDL receptor mediated LDL uptake in HepG2 cells, confirming that the fusion of the HaloTag to the C-terminal of PCSK9 did not affect the function of PCSK9. The potency of PCSK9-HT was lower when compared to the non-tagged wild type recombinant PCSK9. This effect may be the results of the protein's partial purity.

Two main features of HaloTag ligands are their spectral differences and their different cell permeability. We utilized these features by first labeling the 14-13-E4 cells overnight with the impermeable ligand, Alexa Fluor 488, to bind all the secreted PCSK9 in the cell media. The same group of cells was then labeled with cell permeable ligand, TMR, for 15 min. Through the sequential labeling, we were able to differentiate the two pools of PCSK9: the newly synthesized intracellular pool and the internalized pool following secretion (Fig. 1C). The two pools of PCSK9 have shown a distinct pattern of staining. The PCSK9 staining of intracellular pool was predominantly uniform whereas the internalized pool displayed a characteristic punctate staining. The ability to separate the two pools of PCSK9 utilizing different HaloTag ligands enabled us to further study the trafficking and fate of PCSK9 in 14-13-E4 cells.
Subcellular Localization of Intracellular and Internalized PCSK9 is Different
Next, we investigated the trafficking patterns of two pools of PCSK9 in 14-13-E4 cells. The experimental design is shown in Fig. (2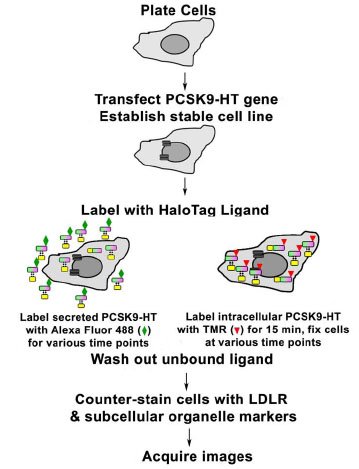 ). To monitor the trafficking of the intracellular pool of PCSK9, 14-13-E4 cells were labeled with 5 mM TMR for 15 min. To allow protein trafficking, the unbound ligand was washed off and the cells were incubated in fresh DMEM (supplemented with 10% FBS) at 37°C + 5% CO2 from 30 to 300 min. The process was stopped by fixation and permeabilization of cells at various time points. The localization of PCSK9 was determined by counterstaining the cells with different subcellular organelle markers, including PDI (ER), Golgin97 (Golgi complex), EEA1 (early endosome), and LAMP1 (lysosome). As shown in Fig. (3
). To monitor the trafficking of the intracellular pool of PCSK9, 14-13-E4 cells were labeled with 5 mM TMR for 15 min. To allow protein trafficking, the unbound ligand was washed off and the cells were incubated in fresh DMEM (supplemented with 10% FBS) at 37°C + 5% CO2 from 30 to 300 min. The process was stopped by fixation and permeabilization of cells at various time points. The localization of PCSK9 was determined by counterstaining the cells with different subcellular organelle markers, including PDI (ER), Golgin97 (Golgi complex), EEA1 (early endosome), and LAMP1 (lysosome). As shown in Fig. (3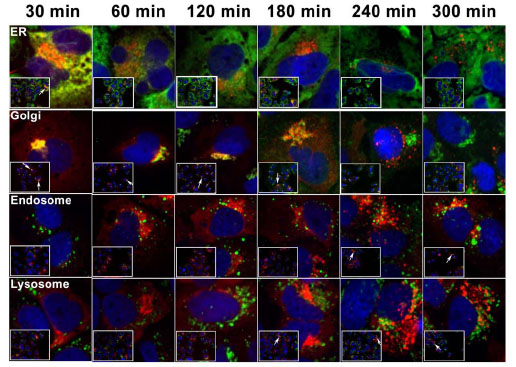 ), intracellular PCSK9 was primarily located in the ER and Golgi after 30 min of labeling and subsequently moved mostly to Golgi locations (up to 180 min post-labeling). The localization of PCSK9 was eventually observed in lysosomes (180-300 min post-labeling). Localization of intracellular PCSK9 with early endosomes, noted at 240 min and 300 min of incubation, was observed as well. These endosome/lysosome localized TMR labeled PCSK9 molecules may have transferred to the endosome/lysosome directly from the Golgi, or these proteins may have been secreted out before re-internalized during incubation period.
), intracellular PCSK9 was primarily located in the ER and Golgi after 30 min of labeling and subsequently moved mostly to Golgi locations (up to 180 min post-labeling). The localization of PCSK9 was eventually observed in lysosomes (180-300 min post-labeling). Localization of intracellular PCSK9 with early endosomes, noted at 240 min and 300 min of incubation, was observed as well. These endosome/lysosome localized TMR labeled PCSK9 molecules may have transferred to the endosome/lysosome directly from the Golgi, or these proteins may have been secreted out before re-internalized during incubation period.


A different strategy of labeling was used to track the internalized PCSK9 pool (Fig. 2). The 14-13-E4 cells were labeled with Alexa Fluor 488 for various time points, ranging from 30 to 300 min and including overnight incubation, followed by fixation and permeabilization. The same set of markers for subcellular organelles as employed for the intracellular PCSK9 pool was applied to locate the Alexa Fluor 488 labeled internalized PCSK9. The localization of the internalized PCSK9 pool was distinct from that of the intracellular PCSK9 (Fig. 4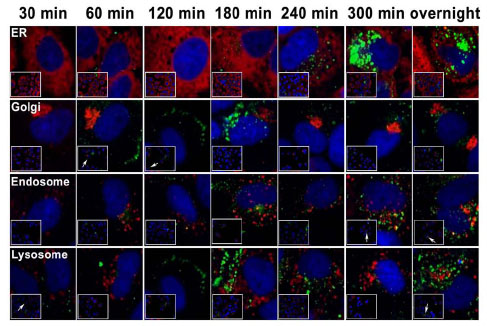 ). Alexa Fluor 488 labeled PCSK9 appeared on the cell surface shortly after labeling (Fig. 4 and Supplement movie 1). Trafficking of the internalized PCSK9 from the cell membrane to the endosome/lysosome was observed after 300 min to overnight labeling of secreted PCSK9. The lack of localization of the internalized PCSK9 in the ER or Golgi further confirmed that the labeling was solely on secreted PCSK9.
). Alexa Fluor 488 labeled PCSK9 appeared on the cell surface shortly after labeling (Fig. 4 and Supplement movie 1). Trafficking of the internalized PCSK9 from the cell membrane to the endosome/lysosome was observed after 300 min to overnight labeling of secreted PCSK9. The lack of localization of the internalized PCSK9 in the ER or Golgi further confirmed that the labeling was solely on secreted PCSK9.

Co-localization of LDL Receptor with the Two Pools of PCSK9-HT
PCSK9 has become an appealing target for the treatment of dyslipidemia due to the findings that it binds the LDL receptor (LDLR) thus affecting LDL uptake [11]. Secreted PCSK9 can bind the LDLR on the cell surface. The PCSK9-LDLR complex is internalized into endosome and degraded in the lysosome. It has also been claimed that newly synthesized PCSK9 may bind LDLR prior to secretion, therefore leading to direct LDLR degradation in the lysosome [19Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment Proc Natl Acad Sci USA 2005; 102: 2069-74.-21Poirier S, Mayer G, Poupon V, et al. Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation evidence for an intracellular route J Biol Chem 2009; 284: 28856-64.]. However, this proposed "PCSK9 intracellular pathway" was established with an over-expression system and was not validated in an in vivo setting. To shed light on PCSK9 trafficking through these two pathways, we examined co-localization of LDLR with these two pools of PCSK9 using HaloTag technology. The experiments were carried out as described above (Fig. 2). Cells with a TMR labeled intracellular pool of PCSK9 or an Alexa Fluor 488 labeled internalized pool of PCSK9 were counterstained with antibodies against LDLR as well as different markers for sub-cellular organelles at various time points. Consistent with the reported results, the secreted PCSK9 co-localized with LDLR in the early endosome and lysosome (Fig. 5A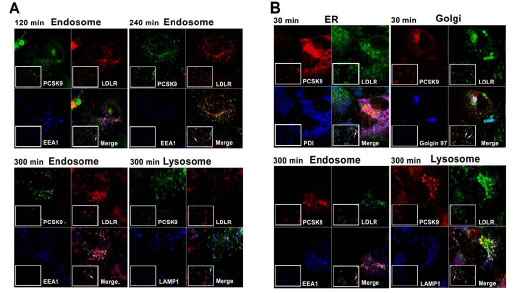 ). Co-localization of PCSK9 and LDLR appeared in the early endosome at about 120 min of Alexa Fluor 488 labeling, and continued to show endosome co-localization at later time points as well. Meanwhile lysosome co-localization was observed after longer incubation time at 300 min of Alexa Fluor 488 labeling, which agrees with a trafficking pattern of the PCSK9-LDLR complex from early endosomes to lysosomes. For the TMR labeled intracellular pool of PCSK9, co-localization with LDLR was observed in the ER and Golgi at 30 min post-labeling. Similar to Alexa Fluor 488 labeling, co-localization of PCSK9 and LDLR in the early endosome and lysosome was also observed at a later time point at about 300 min post-labeling (Fig. 5B). In the current experimental design, however, we were not able to differentiate whether the TMR labeled PCSK9 located in early endosome/lysosome had been secreted prior to entering the early endosome or lysosome. Further investigation is needed to address this question.
). Co-localization of PCSK9 and LDLR appeared in the early endosome at about 120 min of Alexa Fluor 488 labeling, and continued to show endosome co-localization at later time points as well. Meanwhile lysosome co-localization was observed after longer incubation time at 300 min of Alexa Fluor 488 labeling, which agrees with a trafficking pattern of the PCSK9-LDLR complex from early endosomes to lysosomes. For the TMR labeled intracellular pool of PCSK9, co-localization with LDLR was observed in the ER and Golgi at 30 min post-labeling. Similar to Alexa Fluor 488 labeling, co-localization of PCSK9 and LDLR in the early endosome and lysosome was also observed at a later time point at about 300 min post-labeling (Fig. 5B). In the current experimental design, however, we were not able to differentiate whether the TMR labeled PCSK9 located in early endosome/lysosome had been secreted prior to entering the early endosome or lysosome. Further investigation is needed to address this question.
 |
Fig. (5) Co-localization of PCSK9 with LDLR. A. 14-13-E4 cells were labeled with 0.2 µM Alexa Fluor 488 (Green) as in Fig. (4 |
DISCUSSION
The expression of HaloTag fusion PCSK9 (PCSK9-HT) is a key tool in this study to investigate the fate of PCSK9. We based the study on the premise that in any cell the PCSK9 (and PCSK9-HT) can only originate from one of two sources: either the cell synthesizes new protein (translated and expressed de novo), which will be trafficked throughout the cell and (at least partially) secreted, or the cell takes up external PCSK9, which has been previously secreted by any cell. Based on this belief, we utilized the features of the HaloTag ligands to distinguish the source of PCSK9-HT while studying its distribution. We selected a cell line with a PCSK9-HT expression level close to the endogenous level (~ 2 fold) of PCSK9 (Fig. 1A). This strategy should help avoid any artifacts caused by over-expression of a recombinant protein.
The various HaloTag ligands covalently bind the HaloTag in a one to one stoichiometry, i.e. an individual HaloTag (and thereby the PCSK9 molecule fused with it) will remain intact with the first label bound to it throughout the remainder of the experimental procedure. Since each label can be restricted to PCSK9-HT of one origin, it will only represent molecules of that particular origin throughout the experiment.
In this study, we utilized the cell-impermeant Alexa Fluor 488 HT ligand (green fluorescence emission) and the cell-permeant TMR HT ligand (red fluorescence emission). If cell permeant TMR HT ligand is added to the cells after wash-out of extracellular secreted PCSK9-HT, then all intracellular PCSK9 will be labeled. One caveat is that the origin of the PCSK9-HT labeled this way is not clear, since previously internalized PCSK9 is also subject to TMR staining. Alternatively, if cell impermeant Alexa Fluor 488 HT ligand is added to cells in PCSK9-HT containing media (secreted by the cells or added after purification), only the PCSK9 outside the cells will be labeled. If the ligand concentration is significantly higher than the PCSK9-HT concentration, almost complete saturation of PCSK9-HT with Alexa Fluor 488 ligand can be achieved. We postulate that any Alexa Fluor 488 labeled PCSK9-HT detected inside a cell has to be the result of PCSK9 uptake, presumably via LDLR internalization. Through sequential addition of the HT ligands at key time-points, the relationship between intra- and extra-cellular PCSK9 has thus been evaluated (Fig. 1C).
Even though the function of circulating PCSK9 in accelerating the degradation of the LDLR has been well established, two critical questions are still under investigation: (1) The exact mechanism by which PCSK9 causes the LDLR to be targeted for degradation is poorly understood, and (2) It is still unknown whether only external PCSK9 taken up into the cell after LDLR binding can serve this function [11]. The answer to the second question will require a detailed understanding of the contact points between newly synthesized PCSK9 and LDLR in the cell. This study was designed to provide basic data for that purpose.
Supplemental movies 1 and 2 displayed the same cell. In movie 1, the green channel representing the cell impermeant Alexa Fluor 488 ligand is shown as prevalent color, whereas in movie 2, the red channel representing the cell permeant TMR ligand is emphasized. The translocation of the green signal from the membrane, and the occasional formation of vesicles in the focal plane, can be observed. The red signal shows a very different distribution, emphasizing the difference in the cellular fate of the intracellular vs. internalized PCSK9.
As predicted, PCSK9-HT labeled outside the cell with cell-impermeant Alexa Fluor 488 ligand was taken up into the cell and could be detected on the cell surface, followed by the endosome and lysosome, where it co-localized with the LDLR. Low or null level of Alexa Fluor 488 signal was detected in the ER and Golgi (Figs. 4, 5A). This phenomenon correlates well with the known pathway of PCSK9 co-internalization with the LDLR, followed by trafficking through the endosome to the lysosome for degradation. Addition of cell permeant TMR HT ligand yielded a different staining pattern. The label was predominantly in the ER and Golgi at the early time points post-labeling, i.e. the TMR ligand bound PCSK9-HT which was inside the protein synthesis and maturation pathway (Figs. 3, 5B). At the later time points, TMR signal was observed in endosomes and lysosomes; however, we have not yet been able to distinguish whether this is due to re-uptake or direct intracellular trafficking. Therefore, further studies are required to determine whether PCSK9 mediated LDLR degradation occurs exclusively via the internalization pathway (i.e. intracellularly synthesized PCSK9 is secreted and subsequently endocytosed in conjunction with LDLR) or prefers an intracellular pathway. The model system we have developed allows us to next address this in combination with tools to block cellular uptake, possibly utilizing for example dynasore [24] or knock down of ARH [16].
CONCLUSION
Distinguishing the source and fate of a protein in living cells is a long desired tool for cell biology and mechanism of action studies. In this report, we demonstrated that the HaloTag technology affords this ability to researchers based on the different cell permeability and fluorescent spectra of the HaloTag ligands. Utilizing this technology, we learned that the origin of PCSK9-HT protein, either intracellular or extracellular, showed distinct subcellular localization patterns, which might be the basis for diverse PCSK9 actions on LDLR.
CONFLICT OF INTEREST
The authors are employees of Merck and Co., Inc.
ACKNOWLEDGEMENTS
The authors thank Dr. Mei Cong for her expert advice and technical assistance on HaloTag technology; Dr. Douglas Johns for his valuable and critical comments on the manuscript; Ms. Cheryl Schwartz for her kind help on the grammatical proof-reading.