- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Arthritis Journal
(Discontinued)
ISSN: 1876-5394 ― Volume 7, 2014
Disruption of Rankl/Rank Signaling Reduces TNF-Induced Joint Inflammation In Vivo
Zhenqiang Yao1, Ping Li2, Qian Zhang1, Ruolin Guo1, Edward M. Schwarz2, Brendan F. Boyce1, 2, Lianping Xing*, 1, 2
Abstract
TNF is a dominant inflammatory cytokine that is over-produced in the joints of patients with rheumatoid arthritis, as well as animal models of this disease. It promotes bone destruction and inflammation through stimulation of osteoclast formation and activation. While it has been established that the RANKL/RANK system is essential for TNF-induced osteoclast formation and bone erosion in vivo, its role in TNF-induced inflammation remains poorly understood and controversial. Here, we report that genetic and biologic disruption of the RANKL/RANK signaling eliminates knee joint inflammation in TNF-Tg/RANKL-/-, TNF-Tg/RANK-/- and RANK:Fc treated-TNF-Tg mice as determined by functional assessments and histomorphometry. Among several potential mechanisms that could be responsible for this decrease in inflammation, we found that RANKL synergizes with TNF to stimulate TNF production by osteoclast precursors. Thus, blockade of RANKL/RANK signaling may be a viable therapeutic strategy to prevent inflammation and bone loss in diseases where elevated TNF is a key pathogenic factor.
Article Information
Identifiers and Pagination:
Year: 2009Volume: 2
First Page: 7
Last Page: 13
Publisher Id: TOARTHJ-2-7
DOI: 10.2174/1876539400902010007
Article History:
Received Date: 31/12/2008Revision Received Date: 27/1/2009
Acceptance Date: 29/1/2009
Electronic publication date: 26/2/2009
Collection year: 2009
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http: //creativecommons.org/licenses/by-nc/3.0/ which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Department of Pathology and Laboratory Medicine, University of Rochester Medical Center, 601 Elmwood Avenue, Box 626, Rochester, New York, 14642, USA; Tel: 585-273-4090; Fax: 585-756-4468; E-mail: lianping_xing@urmc.rochester.edu
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 31-12-2008 |
Original Manuscript | Disruption of Rankl/Rank Signaling Reduces TNF-Induced Joint Inflammation In Vivo | |
INTRODUCTION
The receptor activator of NF-êB ligand (RANKL)/ RANK system is essential for osteoclast formation. RANKL was initially characterized as a T-lymphocyte-specific protein [1], and subsequently was detected in bone marrow stromal cells and osteoblasts [2, 3]. Later studies demonstrated that RANKL is produced by almost all cell types in the body, including endothelial cells and chondrocytes [4]. Its expression by these cells increases in response to a variety of factors, including cytokines, growth factors and hormones to induce osteoclast formation, activation and survival in normal and disease states [5-7]. In contrast to the broad expression of RANKL, RANK, the receptor for RANKL, is only identified on mature osteoclasts, dendritic cells, and precursor cells of osteoclasts and dendritic cells [4, 8, 9], which are therefore, the major target cells for RANKL. RANK expressing cells appear not to produce RANKL, thus the signal is transduced through a paracrine mechanism.
Despite the essential role of RANKL in the differentiation of osteoclast precursors (OCPs) to mature osteoclasts, little is known as to whether or not it affects other functions of these cells. Recently, several studies have demonstrated that RANKL treatment stimulates OCPs or osteoclasts to produce various cytokines and chemokines, including IL-1, TNF and MCP-1 [10-13], indicating that the RANKL/RANK system is not only critical for osteoclastogenesis to resorb bones, but also important for their activation and effector cell function.
The importance of this RANKL signal in osteoclasts and OCPs towards their activation and contribution to the pathogenesis of inflammatory arthritis has not been addressed because earlier studies failed to detect effects on inflammation. For example, osteoprotegerin (OPG):Fc treatment prevented bone loss and cartilage destruction, but had no discernible effect on inflammation in adjuvant-induced arthritis in Lewis rats [14]. In serum transfer induced arthritis, RANKL-/- mice develop a comparable degree of inflammation to wild type (wt) littermates, but bone erosion was greatly reduced [15]. Thus, the current paradigm is that RANKL-mediated OCP differentiation is critical for bone erosion, but inflammation is either independent of RANKL signaling, or is mediated by several signaling pathways which have overlapping functions in vivo.
During the course of studying the role of the RANKL/ RANK signaling in TNF-mediated osteoclastogenesis in vivo, we generated TNF-Tg/RANK-/- hybrid mice. To our surprise, these mice not only had no bone erosion [16], but also had no or slight inflammation in their knee joints. To further elucidate the effect of RANKL blockade on TNF-induced arthritis and the mechanisms involved, we generated TNF-Tg/RANKL-/- mice and found that they also had little to no knee joint inflammation. More importantly, RANK:Fc treatment of TNF-Tg mice also significantly reduced the severity of their knee joint inflammation. In vitro, we found that RANKL and TNF synergistically stimulate TNF production by OCPs, indicating that blockade of the RANKL/ RANK pathway may prevent joint inflammation in TNF-induced arthritis through inhibition of OCP activation.
EXPERIMENTAL PROCEDURES
Animals
The 3647 TNF-Tg were originally obtained from Dr. G. Kollias [17]. TNF-Tg/RANK-/- mice were characterized previously [16, 18]. RANKL-/- mice in a C57Bl/6 background were provided Dr. S.C. Marks [19] and used to produce the TNF-Tg/RANKL-/- mice through mating. KRN-TCR-Tg mice were obtained from Drs. Mathis and C. Benoist [20]. K/BxN mice were generated by breeding KRN-TCR-Tg mice with nonobese diabetic mice (The Jackson Laboratory, Bar Harbor, ME). All animal studies were approved by the Institutional Animal Care and Use Committee of Rochester University.
Arthritis Models in RANKL Blockade Background and Clinical Assessment
Three models were used. 1) TNF-Tg/RANKL-/- mice were generated by intercrossing TNF-Tg and RANKL+/- mice. Clinical evaluation was performed once a week, starting at 10 week of age in TNF-Tg (N=21) and TNF-Tg/RANKL-/- (N=5) mice for 8 weeks. Paw swelling and grip strength scores were evaluated as reported previously [21]. The deformation score was used because we observed that the first easily recognized sign in TNF-Tg mice is joint deformation, including toe flexion, contraction and shortening. The severity of joint deformation is defined as 4 scores: 0-no deformation of toes and ankle, 1-mild, 2-moderate, 3-severe deformation. 2) Eight-week-old RANK-/- mice and RANK+/- littermates (N=3 per genotype) were injected with serum obtained from arthritic K/BxN mice (10 μl/gram of body weight, i.p.) on day 0 and day 2. Paw swelling was observed once a day for 3 weeks. 3) Ten-week-old TNF-Tg mice (N=8 per group) were given RANK:Fc (10 mg/kg, i.p.) or vehicle three time a week for 8 weeks. Leg bones were harvested for histology and blood was used for measurement of hTNF levels by the end of experiments.
Histology
Paraffin sections were stained with H&E or for TRAP activity. Histomorphometric analysis were performed using an OsteoMeasure program (Osteometrics, Atlanta, GA) [18]. Peri-articular inflammation (mm2) and eroded articular surface (%) were measured around knee joints and the number of osteoclast per millimeter bone surface was countered in the proximal tibiae region.
Osteoclast Precursor Cultures and Quantitative Real-Time RT-PCR
Splenocytes from WT and RANK-/- mice were cultured with M-CSF for 3 days to generate OCPs as described previously [22]. Cells were treated with various amounts of RANKL, TNF or in combination; and the murine TNF and RANKL mRNA expression levels were determined by real time RT-PCR analysis as described previously [22].
Statistical Analysis
Data are shown as mean ± SEM. Group mean values were compared by the Mann–Whitney U-test for non-Normal distributed data and ANOVA flowed by student t-test for normal distributed data.
RESULTS AND DISCUSSION
RANKL Blockade Prevents Joint Inflammation of TNF-Tg Mice
To explore the possibility that RANKL/RANK signaling is involved in arthritis, we compared joint phenotypes of TNF-Tg, RANKL-/-, TNF-Tg/RANKL-/- and wt mice. The gross appearance, movement, and strength of TNF-Tg/RANKL-/- mice were similar to that of RANKL-/-mice, but markedly improved compared to TNF-Tg mice (Fig. 1A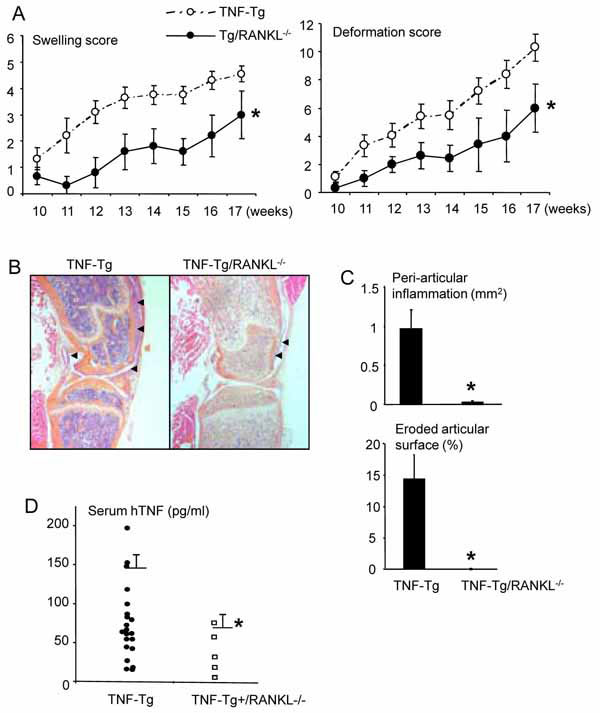 ). As expected, all TNF-Tg mice (21/21) generated from the same mating developed joint inflammation that was readily apparent from histology (Fig. 1B). Also as predicted, all TNF-Tg/RANKL-/- mice (5/5) develop severe osteopetrosis due to a lack of TRAP+ osteoclast formation (Fig. 1B). However, 4 of 5 TNF-Tg/RANKL-/- mice had no knee joint inflammation and their joint structures were the same as RANKL-/- mice by histology (Fig. 1C). One TNF-Tg/ RANKL-/- mouse had a slightly thinker synovium (Fig. 1B). The serum hTNF levels in TNF-Tg/RANKL-/- mice were significantly lower than those of TNF-Tg mice (Fig. 1D). Murine TNF levels from blood of these mice were undetectable using a commercial available TNF ELISA kit [18].
). As expected, all TNF-Tg mice (21/21) generated from the same mating developed joint inflammation that was readily apparent from histology (Fig. 1B). Also as predicted, all TNF-Tg/RANKL-/- mice (5/5) develop severe osteopetrosis due to a lack of TRAP+ osteoclast formation (Fig. 1B). However, 4 of 5 TNF-Tg/RANKL-/- mice had no knee joint inflammation and their joint structures were the same as RANKL-/- mice by histology (Fig. 1C). One TNF-Tg/ RANKL-/- mouse had a slightly thinker synovium (Fig. 1B). The serum hTNF levels in TNF-Tg/RANKL-/- mice were significantly lower than those of TNF-Tg mice (Fig. 1D). Murine TNF levels from blood of these mice were undetectable using a commercial available TNF ELISA kit [18].

To confirm the role of RANKL/RANK signaling in TNF-induced inflammation in an independent genetic model and in non-osteopetrotic mice, we analyzed bone sections from 4-month-old TNF-Tg/RANK-/- mice (N=3) as well as from RANK:Fc treated TNF-Tg mice (N= 8), which received RANK:Fc for 8 weeks from 10-week-old when the arthritis like symptoms just begin. Similar to TNF-Tg/RANKL-/- mice, TNF-Tg/RANK-/- mice had neither joint inflammation nor bone erosion. RANK:Fc treatment significantly decreased the inflammatory area in the knee joints of all 8 TNF-Tg mice, compared to vehicle-treated TNF-Tg mice. Similar to TNF-Tg/RANKL-/- mice, RANK-Fc treated mice had decreased serum hTNF levels than PBS-treated mice (Fig. 2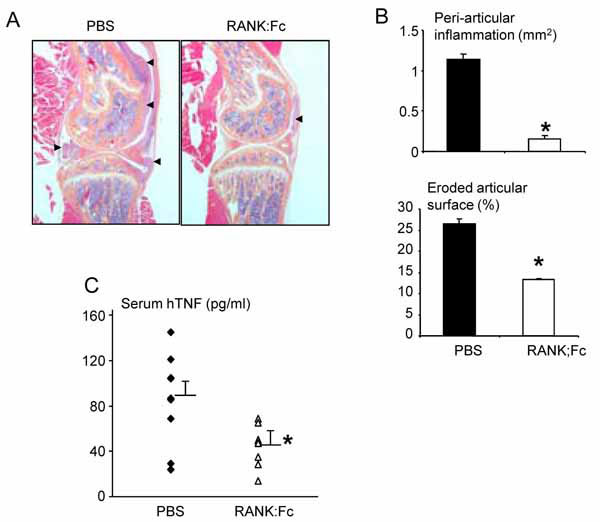 ). Together, our data definitively show that genetic disruption of RANKL signaling from birth abolishes, while inhibition of RANKL at the onset of inflammatory arthritis significantly diminishes TNF-induced joint inflammation.
). Together, our data definitively show that genetic disruption of RANKL signaling from birth abolishes, while inhibition of RANKL at the onset of inflammatory arthritis significantly diminishes TNF-induced joint inflammation.
 |
Fig. (2) RANK:Fc-treated TNF-Tg mice have reduced synovial inflammation in joints. Ten-week-old TNF-Tg were treated with
RANK:Fc or PBS for 2 months. (A) Representatives of H&E-stained sections from a PBS-treated TNF-Tg and a RANK:Fc-treated TNF-Tg
mouse (2X magnification). The area of synovial tissue was indicated by black arrow heads. (B) Histomorphometric analyses of knee joint
sections and (C) serum human TNF transgene concentration were measured as described in Fig. (1 |
Since our findings differ from previous publications in which the blockade of RANKL signaling pathway by OPG:Fc or the induction of arthritis by serum transfer in RANKL-/- mice does not prevent joint inflammation [14, 15, 23], we used another model to test if RANKL blockade only benefits TNF-induced arthritis. In these experiments we induced arthritis by injecting arthritic serum from K/BxN mice into RANK-/- or RANK+/- mice, and measured paw thickness daily starting one day after the last serum injection. As reported in RANKL-/- mice using the same model [15], both RANK-/- and RANK+/- mice developed a similar degree of joint redness and swelling (data not shown). Histomorphometric analysis of the ankle joints of these mice confirmed the similar severity of inflammation (mean +/- SEM of the percentage of inflammation area in knee joints: 5.6+/-1.16 in RANK+/- mice vs 7.7+/-0.34 in RANK-/- mice, N=3 mice per genotype, p>0.05). Thus, the RANKL/RANK dependent effects on inflammation appear to be selective for TNF-induced arthritis.
RANKL Synergistically with TNF to Stimulate TNF Production by OCPs
To elucidate the mechanisms by which RANKL blockade inhibits inflammation specifically in TNF-induced arthritis, we reasoned that RANKL must affect a factor that is essential for the initiation of joint inflammation in TNF-induced, but not for the serum-induced arthritis, and that this factor must be produced by RANK expressing cells. We focused on TNF because it is the driving force for the onset of inflammatory arthritis in TNF-Tg mice [17], but not for mice receiving arthritic serum [24].
TNF is produced by many cell types at inflammatory foci, including macrophages, synoviocytes, T cells, dendritic cells, osteoclasts and OCPs [25]. Among these, only dendritic cells, osteoclasts and OCPs are RANK expressing cells. Since TNF-Tg/c-Fos-/- mice have no osteoclasts, but have elevated serum TNF levels and joint inflammation [21], it is likely that the absence of inflammation in TNF-Tg/RANK-/- or TNF-Tg/RANKL-/- mice is not due to a lack of mature osteoclast-produced TNF. To examine if RANKL promotes TNF production by OCPs that also give rise of dendritic cells, we generated OCPs from splenocytes of wt and RANK-/- mice and treated them with RANKL, TNF or in combination. RANKL synergized with TNF to stimulate TNF production. Importantly, RANKL stimulation at doses 100-fold lower than that required for osteoclast formation, potenicated TNF-induced TNF production (Fig. 3A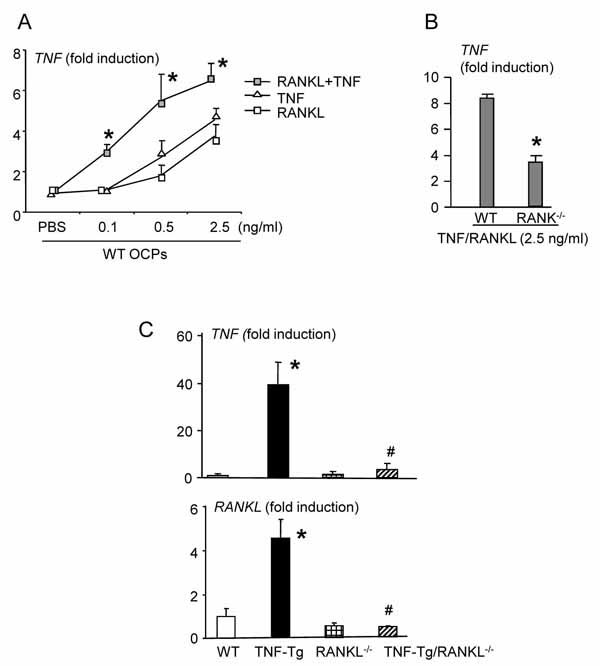 ). This synergistic effect of RANKL and TNF was abolished in RANK-/- cells (Fig. 3B). Thus, a permissive level of RANKL in inflamed joints may be sufficient to stimulate TNF-induced TNF production by OCPs to set-up a positive autocrine loop for inflammation. To test this hypothesis, we examined RANKL and TNF expression levels in the joints of TNF-Tg/RANKL-/- and TNF-Tg mice. RANKL expression was increased in TNF-Tg, but not in TNF-Tg/RANKL-/- mice compared to wt littermates (Fig. 3C). Consistent with a decrease in RANKL mRNA expression, TNF mRNA expression in TNF-Tg/RANKL-/- mice was remarkably reduced (Fig. 3). Interestingly, serum human TNF transgene concentration was decreased in TNF-Tg/RANKL-/- and RANK:Fc-treated TNF-Tg mice compared to control TNF-Tg mice (Figs. 1D and 2C). Currently, the mechanisms by which regulate human TNF transgene expression in TNF-Tg mice are not very clear [17]. Our data suggest that its expression is likely associated to the degree of inflammation.
). This synergistic effect of RANKL and TNF was abolished in RANK-/- cells (Fig. 3B). Thus, a permissive level of RANKL in inflamed joints may be sufficient to stimulate TNF-induced TNF production by OCPs to set-up a positive autocrine loop for inflammation. To test this hypothesis, we examined RANKL and TNF expression levels in the joints of TNF-Tg/RANKL-/- and TNF-Tg mice. RANKL expression was increased in TNF-Tg, but not in TNF-Tg/RANKL-/- mice compared to wt littermates (Fig. 3C). Consistent with a decrease in RANKL mRNA expression, TNF mRNA expression in TNF-Tg/RANKL-/- mice was remarkably reduced (Fig. 3). Interestingly, serum human TNF transgene concentration was decreased in TNF-Tg/RANKL-/- and RANK:Fc-treated TNF-Tg mice compared to control TNF-Tg mice (Figs. 1D and 2C). Currently, the mechanisms by which regulate human TNF transgene expression in TNF-Tg mice are not very clear [17]. Our data suggest that its expression is likely associated to the degree of inflammation.

Based on our in vivo and in vitro findings we propose a model to explain the role of the RANKL/RANK system in TNF-induced arthritis (Fig. 4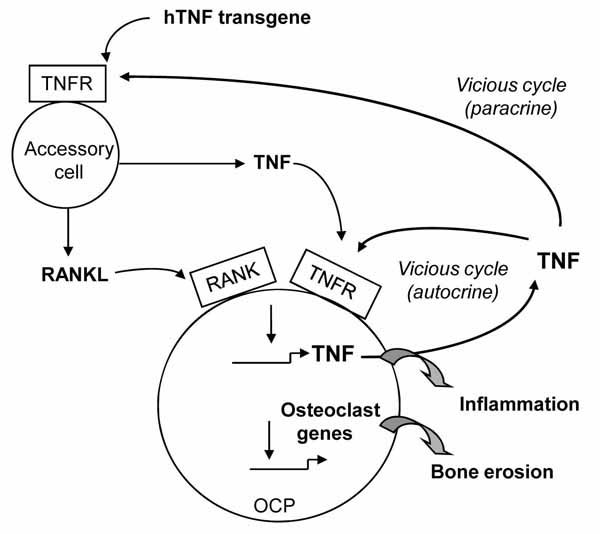 ). In an arthritic joint, overexpressed TNF stimulates accessory cells, including synoviocytes, macrophages and other immune cells, to produce RANKL and TNF. This RANKL and TNF synergistically stimulate RANK+ OCPs to produce more TNF, forming a positive feedback loop, which exacerbates the inflammatory process. In this model, RANKL-mediated TNF auto-regulation plays a critical role in both the initiation and progression of joint disease because if we block the RANKL/RANK pathway before the hTNF transgene level becomes elevated, such as in TNF-Tg/RANKL-/- or TNF-Tg/RANK-/- mice, joint inflammation will not be initiated. However, if RANKL blockade happens after the onset of joint disease at the time when the hTNF transgene levels have been already elevated, such as in RANK:Fc treated 2-month-old TNF-Tg mice, the inflammation still exists but to a much lesser degree. This indicates that RANKL-mediated TNF-auto-regulation also plays a considerable role in the effector phase of the disease.
). In an arthritic joint, overexpressed TNF stimulates accessory cells, including synoviocytes, macrophages and other immune cells, to produce RANKL and TNF. This RANKL and TNF synergistically stimulate RANK+ OCPs to produce more TNF, forming a positive feedback loop, which exacerbates the inflammatory process. In this model, RANKL-mediated TNF auto-regulation plays a critical role in both the initiation and progression of joint disease because if we block the RANKL/RANK pathway before the hTNF transgene level becomes elevated, such as in TNF-Tg/RANKL-/- or TNF-Tg/RANK-/- mice, joint inflammation will not be initiated. However, if RANKL blockade happens after the onset of joint disease at the time when the hTNF transgene levels have been already elevated, such as in RANK:Fc treated 2-month-old TNF-Tg mice, the inflammation still exists but to a much lesser degree. This indicates that RANKL-mediated TNF-auto-regulation also plays a considerable role in the effector phase of the disease.

Our findings raise 2 important issues. One is that RANKL blockade may specifically benefit TNF-induced arthritis or certain forms of inflammatory arthritis where RANK+ cell-produced cytokines plays a critical role. In support of this, a recent study indicates that RANK:Fc treatment significantly reduces the severity of joint inflammation in LPS-induced arthritis [11]. In this report, the authors demonstrate that RANKL stimulated peripheral blood monocytes (PBMC) to produce cytokines and chemokines, although the dose of RANKL used was very high (5 µg/ml), which is probably due to a low percentage of RANK+ cells in PBMC. Whether or not TNF is the initiation factor in this model is not known, but that LPS stimulates TNF production by myeloid cells has been well established [26].
Another issue is how to evaluate results obtained from different animal models of arthritis, because our results strongly argue that using different animal models could result in completely different outcomes in terms of RANKL blockade. This phenomenon is not totally surprising given the variety of mechanisms of actions to induce arthritis in small animals [27]. For instance, K/BxN and adjuvant-induced arthritis is working through auto-antibodies, while arthritis in TNF-Tg or IL-1-Tg mice is through cytokines despite all the models led to joint diseases in the later phase [27]. RANKL blockade is effective in TNF- (our study) or LPS-induced [11], but not in K/BxN serum induced- or adjuvant-induced arthritis [14, 15]. This strongly suggests that the specificity of the initiation factor is critical.
In summary, we have demonstrated a novel role for RANKL/RANK system in mediating cytokine-induced inflammatory arthritis. Since RA pathogenesis is a very complicated process involving autoimmune reactions, cytokine production, and genetic modifications. Anti-RANKL therapy may be a viable therapeutic strategy to prevent inflammation and bone loss in diseases where elevated TNF or other cytokines are the key pathogenic factors.
FOOTNOTES
This work was supported by research grants from the National Institutes of Health (PHS AR 48697 to LX and AR43510 to BFB).
ACKNOWLEDGMENTS
The RANK:Fc used in this study were provided by Amgen Inc. The authors would like to thank Dr. Xiaoyun Zhang for technical assistance with the histology.