- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




The Open Bioactive Compounds Journal
(Discontinued)
ISSN: 1874-8473 ― Volume 9, 2020
Sphingosine Kinase 2 Inhibitor Accelerates Fas-Mediated Cell Death Progression in A20/2J Cells
Miki Hara-Yokoyama*, 1, Kazue Terasawa1, Akio Kihara2, Jin-Wook Kim2, 3, Chan-Seo Park2, 4, Yoshio Hirabayashi5, Yasuyuki Igarashi2 , Masaki Yanagishita1
Abstract
A sphingosine kinase (SPHK) type 2 inhibitor, SG-12, inhibited the SPHK2 activity in A20/2J cells and promoted Fas-dependent cell death. The effect of SG-12 on cell death was relieved by SPHK2 transfection but not by SPHK1 transfection. The results suggest a SPHK2-specific suppression of Fas-dependent cell death.
Article Information
Identifiers and Pagination:
Year: 2008Volume: 1
First Page: 22
Last Page: 27
Publisher Id: TOBCJ-1-22
DOI: 10.2174/1874847300801010022
Article History:
Received Date: 7/8/2008Revision Received Date: 16/9/2008
Acceptance Date: 19/9/2008
Electronic publication date: 28/11/2008
Collection year: 2008
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Department of Hard Tissue Engineering, Biochemistry, Division of Bio-Matrix, Graduate School Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo, Tokyo 113-8549, Japan; Tel: +81 3 5803 5567; Fax: +81 3 5803 0187; E-mail: m.yokoyama.bch@tmd.ac.jp
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 7-8-2008 |
Original Manuscript | Sphingosine Kinase 2 Inhibitor Accelerates Fas-Mediated Cell Death Progression in A20/2J Cells | |
1. INTRODUCTION
Sphingosine kinase (SPHK) catalyzes the phosphorylation of sphingosine to generate sphingosine-1-phosphate (S1P). The two mammalian isoforms of sphingosine kinase, SPHK1 [1Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S. J Biol Chem 1998; 273: 23722-8.
[http://dx.doi.org/10.1074/jbc.273.37.23722] ] and SPHK2 [2Liu H, Sugiura M, Nava VE, et al. J Biol Chem 2000; 275: 19513-20.
[http://dx.doi.org/10.1074/jbc.M002759200] [PMID: 10751414] ], are different in their amino acid sequences, catalytic properties, intracellular localization, and tissue distribution [3Spiegel S, Milstien S. J Biol Chem 2007; 282: 2125-9.]. Mice lacking SPHK1 [4Allende ML, Sasaki T, Kawai H, et al. J Biol Chem 2004; 279: 52487-92.
[http://dx.doi.org/10.1128/MCB.25.24.11113-11121.2005] [PMID: 16314531] [PMCID: PMC1316977] ], SPHK2 [5Kharel Y, Lee S, Snyder AH, et al. J Biol Chem 2005; 280: 36865-72.], or both [6Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Mol Cell Biol 2005; 25: 11113-21.] indicated that the functions of SPHK1 and SPHK2 are redundant in terms of their maintenance of extracellular S1P levels to support S1P-receptor mediated processes such as vascular maturation/angiogenesis [7Wang F, VanBrocklyn JR, Hobson JP, et al. J Biol Chem 1999; 274: 35343-50.
[http://dx.doi.org/10.1074/jbc.274.50.35343] -11Okamoto H, Takuwa N, Yokomizo T, et al. Mol Cell Biol 2000; 20: 9247-61.
[http://dx.doi.org/10.1128/MCB.20.24.9247-9261.2000] [PMID: 11094076] [PMCID: PMC102182] ], heart development [12Kupperman E, An S, Osborne N, Waldron S, Stainier DY. Nature 2000; 406: 192-5.
[http://dx.doi.org/10.1038/35018092] [PMID: 10910360] ], and lymphocyte trafficking [13Graeler M, Shankar G, Goetzl EJ. J Immunol 2002; 169: 4084-7.]. On the other hand, it has been suggested that SPHK1 and SPHK2 have distinct roles in regulating cell survival and death [14Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Biochim Biophys Acta 2006; 1758: 2016-26.], where S1P probably acts intracellularly [14Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Biochim Biophys Acta 2006; 1758: 2016-26.,15Olivera A, Rosenfeldt HM, Bektas M, et al. J Biol Chem 2003; 278: 46452-60.]. Although SPHK1 has been shown to promote cell survival, several reports suggested that SPHK2 induces apoptosis [16Liu H, Toman RE, Goparaju SK, et al. J Biol Chem 2003; 278: 40330-6.
[http://dx.doi.org/10.1074/jbc.M304455200] [PMID: 12835323] ] or inhibits cell growth [17Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S. J Biol Chem 2003; 278: 46832-9.
[http://dx.doi.org/10.1074/jbc.M306577200] [PMID: 12954646] ]. T cells from mice lacking SPHK2 exhibit a hyperactivated phenotype with enhanced proliferation and cytokine secretion in response to IL-2 [18Samy ET, Meyer CA, Caplazi P, et al. J Immunol 2007; 179: 5644-8.]. At present, the role of SPHK2 in modulating cell survival and death remains to be characterized.
Fas/CD95 is a member of the tumor necrosis factor receptor superfamily. Crosslinking Fas with its natural ligand FasL (CD178) or with agonistic antibodies induces apoptotic cell death. Fas signaling is important for elimination of self-reactive T and B lymphocytes [19VanParijs L, Abbas AK. Curr Opin Immunol 1996; 8: 355-61.
[http://dx.doi.org/10.1016/S0952-7915(96)80125-7] , 20Siegel RM, Chan FK, Chun HJ, Lenardo MJ. Nat Immunol 2000; 1: 469-74.
[http://dx.doi.org/10.1038/82712] [PMID: 11101867] ]. Thus, Fas deficiency in humans and mice (e.g., during autoimmune lymphoproliferative syndrome or in lpr mice) predisposes them to systemic autoimmunity. In B cells, susceptibility to Fas signaling is dramatically altered during activation [21Mizuno T, Zhong X, Rothstein TL. Apoptosis 2003; 8: 451-60.
[http://dx.doi.org/10.1023/A:1025534223168] [PMID: 12975576] ].
Sphingolipid ceramide is known to regulate Fas-mediated cell death [22Jarvis WD, Grant S. Curr Opin Oncol 1998; 10: 552-9.
[http://dx.doi.org/10.1097/00001622-199811000-00013] -25Miyaji M, Jin ZX, Yamaoka S, et al. J Exp Med 2005; 202: 249-59.
[http://dx.doi.org/10.1084/jem.20041685] [PMID: 16009715] [PMCID: PMC2213006] ]. Ceramide activates a number of enzymes and contributes to membrane structure, by which it can lead to activation of caspase and induce cell death [26Hannun YA. J Biol Chem 1994; 269: 3125-8., 27Ruvolo PP. Pharmacol Res 2003; 47: 383-92.
[http://dx.doi.org/10.1016/S1043-6618(03)00050-1] ]. On the other hand, the ceramide-derived metabolite S1P, counteracts the action of ceramide and suppresses Fas-mediated cell death [28Cuvillier O, Pirianov G, Kleuser B, et al. Nature 1996; 381: 800-3.
[http://dx.doi.org/10.1038/381800a0] [PMID: 8657285] , 29Bektas M, Jolly PS, Muller C, Eberle J, Spiegel S, Geilen CC. Oncogene 2005; 24: 178-87.
[http://dx.doi.org/10.1038/sj.onc.1208019] [PMID: 15637591] ]. Upregulation of SPHK1 has been reported to induce Fas-resistance in B lymphoblastoid cells in case of rheumatoid arthritis [30Pi X, Tan SY, Hayes M, et al. Arthritis Rheum 2006; 54: 754-64.
[http://dx.doi.org/10.1002/art.21635] [PMID: 16508940] ]. However, the role of SPHK2 in Fas-mediated cell death has not been elucidated.
In the present study, we investigated the role of SPHK on Fas-mediated apoptosis. We found that the SPHK activity in A20/2J cells, a mouse B cell lymphoma cell line, is mainly due to SPHK2. Using a synthetic sphingoid analog, SG-12, previously developed as an SPHK2 inhibitor [31Kim JW, Kim YW, Inagaki Y, et al. Bioorg Med Chem 2005; 13: 3475-85.], we successfully demonstrated the specific involvement of SPHK2 in the suppression of Fas-dependent cell death of A20/2J cells. This effect was especially evident in the presence of BCR-crosslinking.
2. MATERIALS AND METHODS
2.1. Cell Preparation and Culture
Splenic B cells were isolated from six-week-old BALB/cAnN mice as described previously [32Hara-Yokoyama M, Kimura T, Kaku H, et al. Int Immunopharmacol 2008; 8: 59-70.]. The murine B lymphoma-derived cell line A20 and A20/2J were maintained in RPMI 1640 medium (Sigma-Aldrich, St Louis, MO) supplemented with 8% fetal bovine serum (F6178, Sigma-Aldrich, St Louis, MO), 2-mercaptoethanol (50 μM), penicillin (50 U/ml), and streptomycin (50 μg/ml). Only those cells that were in exponential growth phase were used for experiments.
2.2. Reagents
A compound, (2S, 3R)-2-amino-4-(4-octylphenyl)butane-1-3-diol (SG-12), was prepared as previously described [31Kim JW, Kim YW, Inagaki Y, et al. Bioorg Med Chem 2005; 13: 3475-85.] and dissolved in DMSO at 4 mM concentration. Anti-mouse Fas monoclonal antibody (Jo2) and FITC-annexin V were purchased from BD Pharmingen (San Diego, CA). Z-VAD-fmk was from the Peptide Institute (Osaka, Japan).
2.3. Reverse Transcription Polymerase Chain Reaction (PCR)
Total RNA was isolated from B cells, A20, and A20/2J cells using QIAzol reagent (QIAGEN) and cDNA was generated using an oligo(dT) primer and SuperScript II Reverse Transcriptase (Invitrogen). The GAPDH, SPHK1, and SPHK2 cDNAs were amplified by PCR using primers for GAPDH (5’ ACCACAGTCCATGCCATCAC 3’ and 5’ TCCACCACCCCTGTTGCTGTA 3’), SPHK1 (5’ TGCCTTCTCATTGGACTGTG 3’ and 5’ ACCATCAGCTCTCCATCCAC 3’) and SPHK2 (5’ GGCCACCACTTATGAGGAGA 3’ and 5’ AGCAGTTGAGCAACAGGTCA 3’). The PCR conditions were as follows: 30 cycles; 94℃ for 30 s, 60℃ for 45 s, and 72℃ for 60 s.
2.4. Plasmids and Transfection
The pCE-puro 3x FLAG-mSPHK1a and pCE-puro 3x FLAG-mSPHK2 plasmids are derivatives of the pCE-puro vector [33Kihara A, Ikeda M, Kariya Y, Lee EY, Lee YM, Igara-shi Y. J Biol Chem 2003; 278: 14578-85.
[http://dx.doi.org/10.1074/jbc.M211416200] [PMID: 12584204] ] and encode the mouse SPHK1 and SPHK2 proteins, respectively. These are both tagged with a triple FLAG (3x FLAG) epitope at their N-terminus. Transfections were performed using the Nucleofector technology (Amaxa) and puromycin-resistant cells were obtained.
2.5. SPHK Activity
In vitro SPHK assays were performed as described previously [1Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S. J Biol Chem 1998; 273: 23722-8.
[http://dx.doi.org/10.1074/jbc.273.37.23722] , 2Liu H, Sugiura M, Nava VE, et al. J Biol Chem 2000; 275: 19513-20.
[http://dx.doi.org/10.1074/jbc.M002759200] [PMID: 10751414] ]. The SPHK activity was measured in the presence of 0.25% Triton X-100 (inhibiting SPHK2) and 0.2 M KCl (suitable for SPHK2). To evaluate the SPHK activity within cells, A20/2J cells were incubated with [3-3H]sphingosine after incubation with or without SG-12. Lipids were extracted and analyzed as described previously [1Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S. J Biol Chem 1998; 273: 23722-8.
[http://dx.doi.org/10.1074/jbc.273.37.23722] ].
2.6. Flow Cytometric Analysis
A20/2J cells were stained with FITC-annexin V and propidium iodide (PI) and analyzed by flow cytometry using a FACSCalibur (BD Bioscience, Mountain View, CA). Cells were classified as live cells [Annexin V− PI−], apoptotic cells (Annexin V+ PI−), and dead cells (Annexin V+ PI+).
2.7. Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophresis (SDS-PAGE) and Immunoblotting
After stimulation, the A20/2J cells were lysed using 1% Triton X-100 as described previously [32Hara-Yokoyama M, Kimura T, Kaku H, et al. Int Immunopharmacol 2008; 8: 59-70.] in the presence of 100 mM z-VAD-fmk. Proteins were separated by SDS-PAGE on 10-20% gradient gel (Wako Pure Chemical Industries, Japan) and were detected by immunoblotting with anti-caspase 3 (1/2000 dilution), anti-phospho-Akt (Ser473) (1/1000 dilution), and anti-Akt (1/100 dilution) antibodies from Cell Signaling (Beverly, MA) and anti-Grb2 antibody (1/1000 dilution) from Santa Cruz Biotechnology (Santa Cruz, CA) as primary antibodies. A horseradish peroxidase-conjugated antibody was used as the secondary antibody. Finally, immunoreactivity was determined by ECL (Amer-sham International, Buckinghamshire, England).
2.8. Statistical Analysis
Differences between mean values were assessed by a Student’s t test. A p value of <0.05 or <0.01 was considered statistically significant.
3. RESULTS AND DISCUSSION
3.1. Sphingosine Kinase 2 (SPHK2) Mainly Contributed to SPHK Activity in A20/2J Cells
In B cells, the expression of both SPHK1 and SPHK2 was detected (Fig. 1A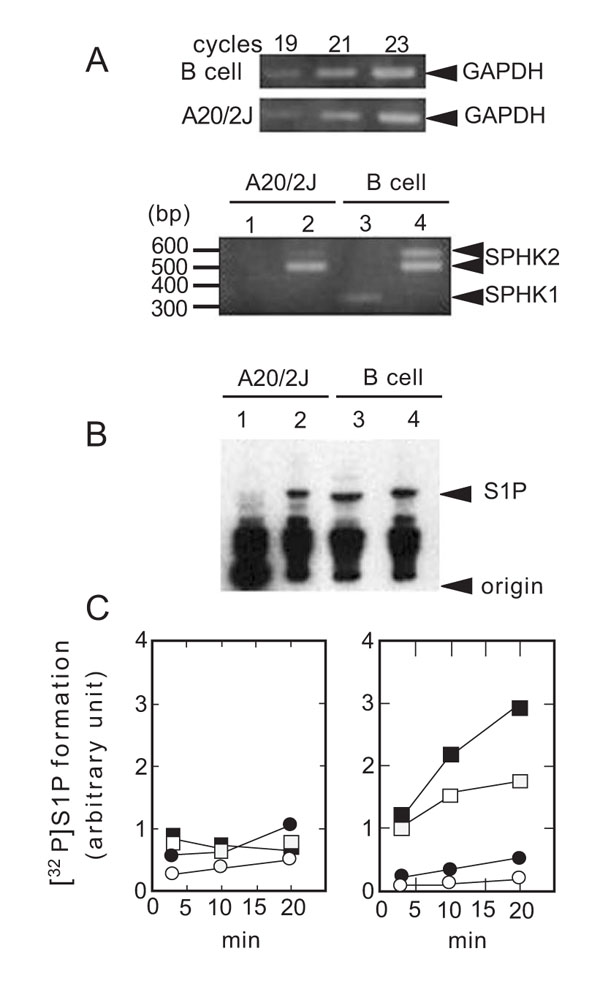 ). In case of SPHK2, two PCR products were observed using B cell cDNA as a template. Sequence analysis revealed that the slower migrating band contained an 83 bp insertion (data not shown). This finding suggested the presence of a splice variant of SPHK2 in B cells. Since a stop codon is present within the insertion, the splice variant would correspond to the N-terminal one third of SPHK2. A role of such truncated form of SPHK2 should be studied in a future work. The B cell tumor line A20/2J is a subclone of the original A20 tumor cell line derived from a BALB/cAnN mouse [34McKean DJ, Infante AJ, Nilson A, et al. J Exp Med 1981; 154: 1419-31.
). In case of SPHK2, two PCR products were observed using B cell cDNA as a template. Sequence analysis revealed that the slower migrating band contained an 83 bp insertion (data not shown). This finding suggested the presence of a splice variant of SPHK2 in B cells. Since a stop codon is present within the insertion, the splice variant would correspond to the N-terminal one third of SPHK2. A role of such truncated form of SPHK2 should be studied in a future work. The B cell tumor line A20/2J is a subclone of the original A20 tumor cell line derived from a BALB/cAnN mouse [34McKean DJ, Infante AJ, Nilson A, et al. J Exp Med 1981; 154: 1419-31.
[http://dx.doi.org/10.1084/jem.154.5.1419] ]. Since A20/2J is more susceptible to Fas-dependent cell death than A20 (data not shown), we used A20/2J for the present study. SPHK2 was clearly observed, whereas that of SPHK1 was barely detectable in A20/2J cells (Fig. 1A) and A20 cells (data not shown).

The catalytic activity of SPHK2 is inhibited by high concentrations of Triton X-100 and activated in the presence of physiological concentrations of KCl or NaCl [2Liu H, Sugiura M, Nava VE, et al. J Biol Chem 2000; 275: 19513-20.
[http://dx.doi.org/10.1074/jbc.M002759200] [PMID: 10751414] ]. Thus, we measured SPHK activity in the presence of 0.25% Triton X-100 (inhibiting SPHK2) and 0.2 M KCl (suitable for SPHK2) to evaluate relative contribution of SPHK2 over that of SPHK1. As shown in Fig. (1B) (lanes 3 and 4), S1P was generated in B cell homogenates under both the conditions. In contrast, S1P formation in the homogenates of A20/2J cells was observed only in the presence of 0.2 M KCl (Fig. 1B, lane 2). SPHK activity under the SPHK2 assay conditions was associated with the membrane fraction (Fig. 1C). Thus, SPHK2 contributed more than SPHK1 to S1P formation in A20/2J cells as compared to B cells.
3.2. A Synthetic Sphingoid Analog Compound (SG-12), (2S, 3R)-2-Amino-4-(4-Octylphenyl)Butane-1-3-Diol, Inhibited SPHK2 Activity in A20/2J Cells
The sphingoid analog, SG-12, potently inhibits human SPHK2 but not SPHK1 activity in vitro [31Kim JW, Kim YW, Inagaki Y, et al. Bioorg Med Chem 2005; 13: 3475-85.]. Since SPHK2 mainly contributed to the SPHK activity in A20/2J cells, we evaluated the action of SG-12 in A20/2J cells. As shown in Fig. (2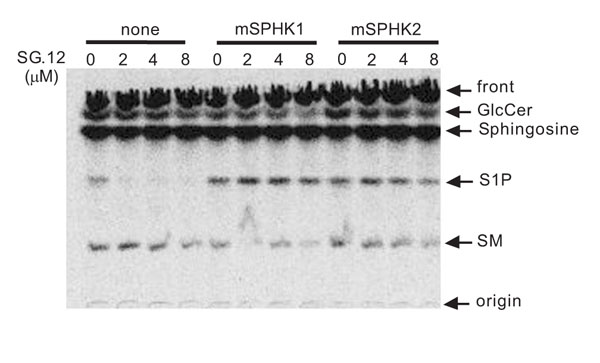 ), SG-12 appreciably inhibited [3H]S1P formation. Thus, SG-12 was shown to act as a membrane-permeable inhibitor of SPHK. To confirm the action of SG-12 on SPHK2, we transfected the A20/2J cells with either mouse SPHK1 or SPHK2. The generation of [3H]S1P was not inhibited by SG-12 in SPHK1-transfected A20/2J cells, but was inhibited in SPHK2-transfected A20/2J cells (Fig. 2). However, the effect of SG-12 in SPHK2-transfected A20/2J cells was less than that in A20/2J cells. Accordingly, the results indicated that SG-12 inhibited the SPHK2 activity in A20/2J cells.
), SG-12 appreciably inhibited [3H]S1P formation. Thus, SG-12 was shown to act as a membrane-permeable inhibitor of SPHK. To confirm the action of SG-12 on SPHK2, we transfected the A20/2J cells with either mouse SPHK1 or SPHK2. The generation of [3H]S1P was not inhibited by SG-12 in SPHK1-transfected A20/2J cells, but was inhibited in SPHK2-transfected A20/2J cells (Fig. 2). However, the effect of SG-12 in SPHK2-transfected A20/2J cells was less than that in A20/2J cells. Accordingly, the results indicated that SG-12 inhibited the SPHK2 activity in A20/2J cells.

3.3. Effect of SG-12 on the Progression of Fas-Mediated Cell Death from Live to Apoptotic Cells
Stimulating A20/2J cells with an anti-mouse Fas monoclonal antibody (Jo2) for 4 h resulted in increase in apoptotic cells (Fig. 3A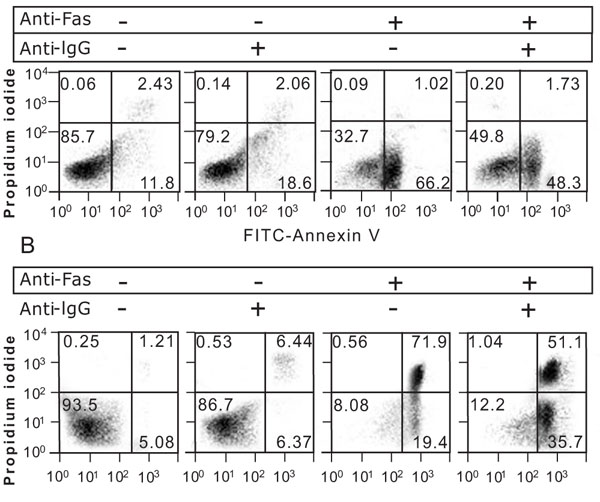 ). Crosslinking of BCR with an anti-mouse IgG pAb reduced the apoptotic cells (Fig. 3A) and inhibited the Fas-dependent activation of caspase-3 (Fig. 4B
). Crosslinking of BCR with an anti-mouse IgG pAb reduced the apoptotic cells (Fig. 3A) and inhibited the Fas-dependent activation of caspase-3 (Fig. 4B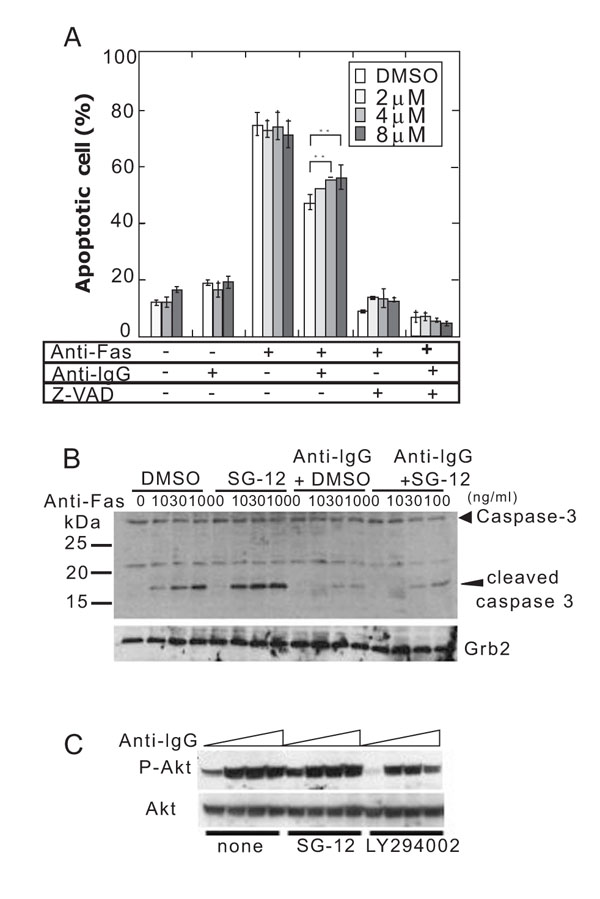 ), which has been known as BCR-dependent Fas-resistance [21Mizuno T, Zhong X, Rothstein TL. Apoptosis 2003; 8: 451-60.
), which has been known as BCR-dependent Fas-resistance [21Mizuno T, Zhong X, Rothstein TL. Apoptosis 2003; 8: 451-60.
[http://dx.doi.org/10.1023/A:1025534223168] [PMID: 12975576] , 35Moriyama H, Yonehara S. Immunol Lett 2007; 109: 36-46.
[http://dx.doi.org/10.1016/j.imlet.2006.12.009] [PMID: 17275920] ]. A proportion of the apoptotic cells was not affected by SG-12 in the absence of BCR-crosslinking, but was increased in the presence of BCR-crosslinking (Fig. 4A). SG-12 did not produce any effects in the presence of a pan-caspase inhibitor, z-VAD-fmk (Fig. 4A), suggesting that SG-12 promoted a caspase-dependent process. Activation of caspase-3 as revealed by the appearance of its cleaved form, was slightly increased by SG-12 either in the absence or presence of BCR-crosslinking (Fig. 4B). Thus, it is suggested that the slight activation of caspase 3 by SG-12 does not directly result in the increase in apoptotic cells. Rather, it implied that SG-12 amplifies an additional process of apoptotic cell death subsequent to the activation of caspase 3, which is evident in the presence of BCR-crosslinking.


The effect of SG-12 could be explained by an impairment of BCR-signaling. However, a BCR-dependent inhibition of caspase 3-activation was clearly observed in the presence of SG-12 (Fig. 3B). In addition, an activation of PI3K as revealed by the phosphorylation of Akt, which mediates the suppressive effect of BCR-crosslinking on Fas-dependent cell death [35Moriyama H, Yonehara S. Immunol Lett 2007; 109: 36-46.
[http://dx.doi.org/10.1016/j.imlet.2006.12.009] [PMID: 17275920] ], was inhibited by LY294002 but not by SG-12 (Fig. 4C). Therefore, the effect of SG-12 in increasing in apoptotic cells may be highlighted when the increase in apoptotic cells is retarded by BCR-crosslinking.
3.4. Effect of SG-12 on the Progression of Fas-Mediated Cell Death from Apoptotic to Dead Cells
Next, we focused on the effect of SG-12 during the late stages of cell death, from visible apoptosis to dead cells. After Fas stimulation for 20 h, most cells became positive for Annexin V and BCR-crosslinking resulted in a decrease in dead cells and an increase in apoptotic cells (Fig. 3B). As shown in Fig. (5A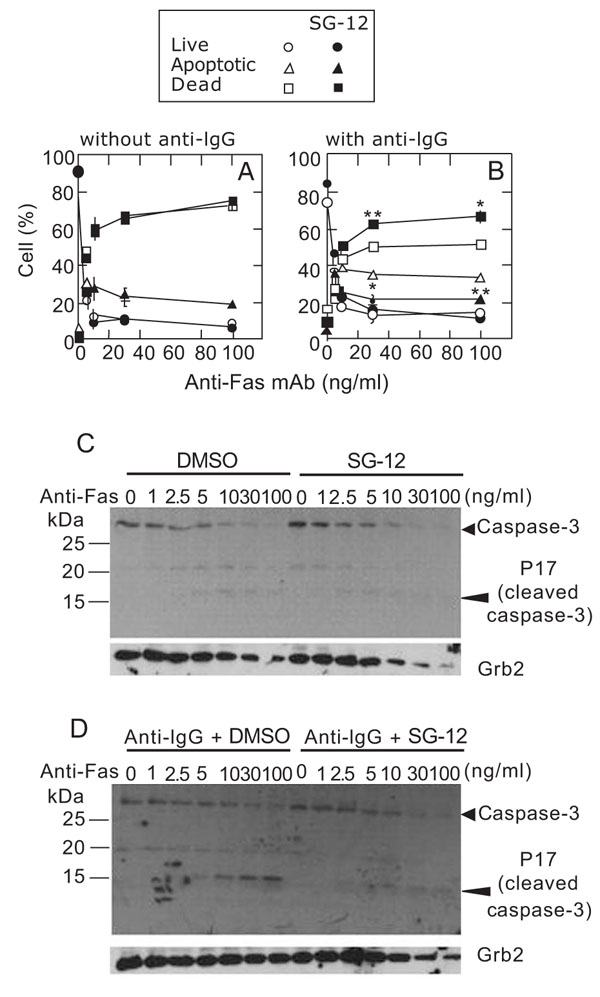 ), the proportions of live, apoptotic, and dead cells were not affected by SG-12 in the absence of BCR-crosslinking. In contrast, the proportion of apoptotic cells was reduced and that of dead cells was increased by SG-12 in the presence of BCR-crosslinking (Fig. 5B). It should be noted that, when the cells were stimulated with an anti-Fas antibody at concentrations higher than 10 ng/ml and in the presence of BCR-crosslinking, the levels of apoptotic and dead cells did not change (at around 35% and 50%, respectively), while the initial activation of caspase 3 was increased (Fig. 5D). This observation suggested that BCR-crosslinking causes a pause in the progression of cells from apoptotic stage to death: addition of SG-12 relieved this pause.
), the proportions of live, apoptotic, and dead cells were not affected by SG-12 in the absence of BCR-crosslinking. In contrast, the proportion of apoptotic cells was reduced and that of dead cells was increased by SG-12 in the presence of BCR-crosslinking (Fig. 5B). It should be noted that, when the cells were stimulated with an anti-Fas antibody at concentrations higher than 10 ng/ml and in the presence of BCR-crosslinking, the levels of apoptotic and dead cells did not change (at around 35% and 50%, respectively), while the initial activation of caspase 3 was increased (Fig. 5D). This observation suggested that BCR-crosslinking causes a pause in the progression of cells from apoptotic stage to death: addition of SG-12 relieved this pause.

An active form of caspase 3 (p17) was hardly detected in the absence of BCR-crosslinking after Fas stimulation for 20 h (Fig. 5C). On the other hand, p17 remained in the presence of BCR-crosslinking; however, it almost disappeared following the addition of SG-12 (Fig. 5D). Since a cytosolic protein, Grb2, was also decreased after Fas-stimulation, it is conceivable that p17 is released from dead cells due to a damage of plasma membranes. It should be noted that SG-12 promoted the decrease in Grb2 both in the absence (Fig. 5C) and presence (Fig. 5D) of BCR-crosslinking, whereas SG-12 did not affect the Fas-dependent decrease in uncleaved caspase 3. It implied that the addition of SG-12 promotes the progression of late stage cell death after activation of caspase 3.
Collectively, SG-12 accelerated both early and late stages of Fas-mediated cell death as shown by the exposure of phosphatidylserine (annexin V-staining) and the change of membrane permeability (PI-staining), respectively.
3.5. Effect of SG-12 on the Progression of Fas-Mediated Cell Death in A20/2J Cells Transfected with SPHK1 or SPHK2
To confirm the involvement of SPHK2 in reactions mediated by SG-12, we used SPHK1- and SPHK2-transfected A20/2J cells (Fig. 6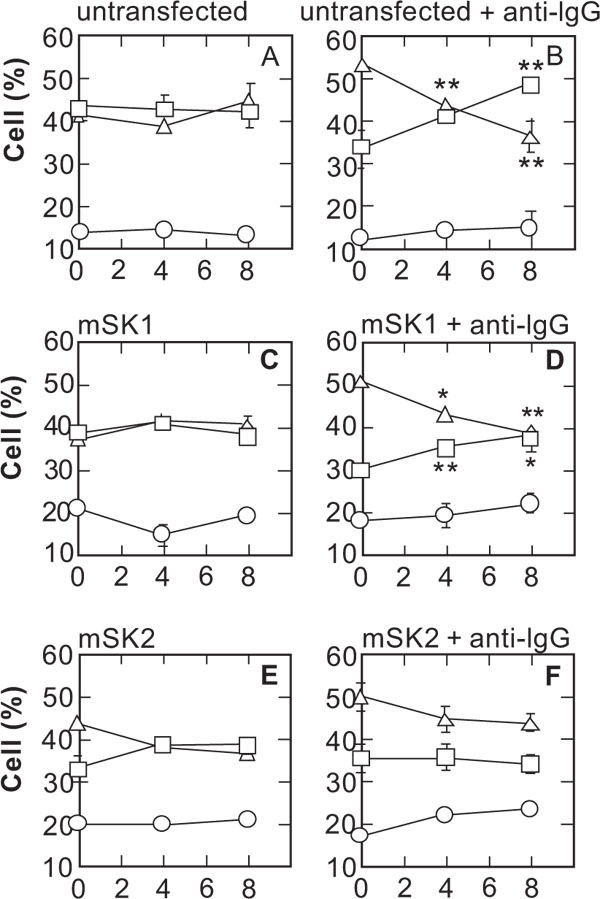 ). SG-12 decreased the apoptotic cells and increased the dead cells after a 20 h Fas stimulation, in the presence of BCR-crosslinking in A20/2J cells (Fig. 6B) and in SPHK1-transfected A20/2J cells (Fig. 6D). In contrast, in SPHK2-transfected A20/2J cells (Fig. 6F), SG-12 did not increase the dead cells. A slight SG-12 mediated decrease in apoptotic cells may reflect an increase of live cells, however, we did not investigate this point further in the present study. The results indicated that the effect of SG-12 in promoting progression of Fas-dependent cell death progression is closely related to SPHK2.
). SG-12 decreased the apoptotic cells and increased the dead cells after a 20 h Fas stimulation, in the presence of BCR-crosslinking in A20/2J cells (Fig. 6B) and in SPHK1-transfected A20/2J cells (Fig. 6D). In contrast, in SPHK2-transfected A20/2J cells (Fig. 6F), SG-12 did not increase the dead cells. A slight SG-12 mediated decrease in apoptotic cells may reflect an increase of live cells, however, we did not investigate this point further in the present study. The results indicated that the effect of SG-12 in promoting progression of Fas-dependent cell death progression is closely related to SPHK2.

4. CONCLUSION
An SK2 inhibitor compound, SG-12, enabled us to demonstrate the involvement of SPHK2 in the suppression of Fas-dependent cell death in A20/2J cells. Interestingly, SG-12 was able to accelerate the progression of late stage cell death indicators such as a change in membrane permeability, probably due to disruption of membrane integrity. This effect was especially evident in the presence of BCR-crosslinking. Although many previous studies focused on the initiation of cell death, i.e., caspase activation, the regulation of the late stages of cell death remains largely unclear. Our results implied the potential ability of SHK2 to suppress the progression of late stage Fas-mediated cell death.
The ability of SG-12 to inhibit S1P formation in A20/2J cells was diminished by SPHK1 transfection (Fig. 2). But SPHK1 transfection did not relieve the effect of SG-12 on the progression of cell death (Fig. 6). Thus, the generation of S1P per se, may not be sufficient to suppress Fas-mediated cell death. On the other hand, it has been previously reported that the generation of ceramide is increased by SPHK2 but decreased by SPHK1 [36Maceyka M, Sankala H, Hait NC, et al. J Biol Chem 2005; 280: 37118-29.
[http://dx.doi.org/10.1074/jbc.M502207200] [PMID: 16118219] ]. SPHK2 probably facilitates the S1P-mediated salvage pathway of ceramide synthesis in which S1P is dephosphorylated to sphingosine by S1P-phosphatase and then acylated to form ceramide in ER [37LeStunff H, Galve-Roperh I, Peterson C, Milstien S, Spiegel S. J Cell Biol 2002; 158: 1039-49.
[http://dx.doi.org/10.1083/jcb.200203123] [PMID: 12235122] [PMCID: PMC2173216] ]. Corroborating previous findings, the generation of the [3H] ceramide-derived metabolites, [3H]GlcCer and [3H]sphingomyelin, in [3H]sphingosine-labeled A20/2J cells were increased by SPHK2 transfection and decreased by SG-12 (Fig. 2). Previously, it has been reported that the generation of ceramide is involved in cell death in B cells [38Kroesen BJ, Pettus B, Luberto C, et al. J Biol Chem 2001; 276: 13606-14., 39Kroesen BJ, Jacobs S, Pettus BJ, et al. J Biol Chem 2003; 278: 14723-31.]. Further study is necessary to correlate such SPHK2-mediated sphingolipid metabolism and the regulation of cell death.
ACKNOWLEDGEMENTS
We thank Dr. Takuma Nakajima and Dr. Katarzyna A. Podyma-Inoue (Tokyo Medical and Dental University) for their valuable discussion. This work was supported by the Grants-in-Aids for 21st Century COE Program to Tokyo Medical and Dental University from the Ministry of Education, Science, Sports and Culture, Japan.
ABBREVIATIONS
| SPHK | = Sphingosine kinase |
| S1P | = Sphingosine-1-phosphate |
| BCR | = B cell receptor |
| SG-12 | = (2S, 3R)-2-amino-4-(4-octylphenyl)butane-1-3-diol |
| PI | = Propidium iodide |
REFERENCES
| [1] | Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S. J Biol Chem 1998; 273: 23722-8. [http://dx.doi.org/10.1074/jbc.273.37.23722] |
| [2] | Liu H, Sugiura M, Nava VE, et al. J Biol Chem 2000; 275: 19513-20. [http://dx.doi.org/10.1074/jbc.M002759200] [PMID: 10751414] |
| [3] | Spiegel S, Milstien S. J Biol Chem 2007; 282: 2125-9. |
| [4] | Allende ML, Sasaki T, Kawai H, et al. J Biol Chem 2004; 279: 52487-92. [http://dx.doi.org/10.1128/MCB.25.24.11113-11121.2005] [PMID: 16314531] [PMCID: PMC1316977] |
| [5] | Kharel Y, Lee S, Snyder AH, et al. J Biol Chem 2005; 280: 36865-72. |
| [6] | Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Mol Cell Biol 2005; 25: 11113-21. |
| [7] | Wang F, VanBrocklyn JR, Hobson JP, et al. J Biol Chem 1999; 274: 35343-50. [http://dx.doi.org/10.1074/jbc.274.50.35343] |
| [8] | Lee MJ, Thangada S, Claffey KP, et al. Cell 1999; 99: 301-12. [http://dx.doi.org/10.1016/S0092-8674(00)81661-X] |
| [9] | English D, Welch Z, Kovala AT, et al. FASEB J 2000; 14: 2255-65. [http://dx.doi.org/10.1096/fj.00-0134com] [PMID: 11053247] |
| [10] | Liu Y, Wada R, Yamashita T, et al. J Clin Invest 2000; 106: 951-61. |
| [11] | Okamoto H, Takuwa N, Yokomizo T, et al. Mol Cell Biol 2000; 20: 9247-61. [http://dx.doi.org/10.1128/MCB.20.24.9247-9261.2000] [PMID: 11094076] [PMCID: PMC102182] |
| [12] | Kupperman E, An S, Osborne N, Waldron S, Stainier DY. Nature 2000; 406: 192-5. [http://dx.doi.org/10.1038/35018092] [PMID: 10910360] |
| [13] | Graeler M, Shankar G, Goetzl EJ. J Immunol 2002; 169: 4084-7. |
| [14] | Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Biochim Biophys Acta 2006; 1758: 2016-26. |
| [15] | Olivera A, Rosenfeldt HM, Bektas M, et al. J Biol Chem 2003; 278: 46452-60. |
| [16] | Liu H, Toman RE, Goparaju SK, et al. J Biol Chem 2003; 278: 40330-6. [http://dx.doi.org/10.1074/jbc.M304455200] [PMID: 12835323] |
| [17] | Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, Nakamura S. J Biol Chem 2003; 278: 46832-9. [http://dx.doi.org/10.1074/jbc.M306577200] [PMID: 12954646] |
| [18] | Samy ET, Meyer CA, Caplazi P, et al. J Immunol 2007; 179: 5644-8. |
| [19] | VanParijs L, Abbas AK. Curr Opin Immunol 1996; 8: 355-61. [http://dx.doi.org/10.1016/S0952-7915(96)80125-7] |
| [20] | Siegel RM, Chan FK, Chun HJ, Lenardo MJ. Nat Immunol 2000; 1: 469-74. [http://dx.doi.org/10.1038/82712] [PMID: 11101867] |
| [21] | Mizuno T, Zhong X, Rothstein TL. Apoptosis 2003; 8: 451-60. [http://dx.doi.org/10.1023/A:1025534223168] [PMID: 12975576] |
| [22] | Jarvis WD, Grant S. Curr Opin Oncol 1998; 10: 552-9. [http://dx.doi.org/10.1097/00001622-199811000-00013] |
| [23] | Watanabe M, Kitano T, Kondo T, et al. Cancer Res 2004; 64: 1000-7. |
| [24] | Grassme H, Cremesti A, Kolesnick R, Gulbins E. Oncogene 2003; 22: 5457-70. [http://dx.doi.org/10.1038/sj.onc.1206540] [PMID: 12934106] |
| [25] | Miyaji M, Jin ZX, Yamaoka S, et al. J Exp Med 2005; 202: 249-59. [http://dx.doi.org/10.1084/jem.20041685] [PMID: 16009715] [PMCID: PMC2213006] |
| [26] | Hannun YA. J Biol Chem 1994; 269: 3125-8. |
| [27] | Ruvolo PP. Pharmacol Res 2003; 47: 383-92. [http://dx.doi.org/10.1016/S1043-6618(03)00050-1] |
| [28] | Cuvillier O, Pirianov G, Kleuser B, et al. Nature 1996; 381: 800-3. [http://dx.doi.org/10.1038/381800a0] [PMID: 8657285] |
| [29] | Bektas M, Jolly PS, Muller C, Eberle J, Spiegel S, Geilen CC. Oncogene 2005; 24: 178-87. [http://dx.doi.org/10.1038/sj.onc.1208019] [PMID: 15637591] |
| [30] | Pi X, Tan SY, Hayes M, et al. Arthritis Rheum 2006; 54: 754-64. [http://dx.doi.org/10.1002/art.21635] [PMID: 16508940] |
| [31] | Kim JW, Kim YW, Inagaki Y, et al. Bioorg Med Chem 2005; 13: 3475-85. |
| [32] | Hara-Yokoyama M, Kimura T, Kaku H, et al. Int Immunopharmacol 2008; 8: 59-70. |
| [33] | Kihara A, Ikeda M, Kariya Y, Lee EY, Lee YM, Igara-shi Y. J Biol Chem 2003; 278: 14578-85. [http://dx.doi.org/10.1074/jbc.M211416200] [PMID: 12584204] |
| [34] | McKean DJ, Infante AJ, Nilson A, et al. J Exp Med 1981; 154: 1419-31. [http://dx.doi.org/10.1084/jem.154.5.1419] |
| [35] | Moriyama H, Yonehara S. Immunol Lett 2007; 109: 36-46. [http://dx.doi.org/10.1016/j.imlet.2006.12.009] [PMID: 17275920] |
| [36] | Maceyka M, Sankala H, Hait NC, et al. J Biol Chem 2005; 280: 37118-29. [http://dx.doi.org/10.1074/jbc.M502207200] [PMID: 16118219] |
| [37] | LeStunff H, Galve-Roperh I, Peterson C, Milstien S, Spiegel S. J Cell Biol 2002; 158: 1039-49. [http://dx.doi.org/10.1083/jcb.200203123] [PMID: 12235122] [PMCID: PMC2173216] |
| [38] | Kroesen BJ, Pettus B, Luberto C, et al. J Biol Chem 2001; 276: 13606-14. |
| [39] | Kroesen BJ, Jacobs S, Pettus BJ, et al. J Biol Chem 2003; 278: 14723-31. |