- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Drug Resistance Journal
(Discontinued)
ISSN: 1876-5211 ― Volume 1, 2011
ERK Suppression in DU145 Cells Selected for AG2034 Resistance is Associated with Decreased ATP, GARFT Inactivation and Increased ERK Signaling
Oluwakemi Obajimi*, Peter W Melera
Abstract
Background:
Previously, we showed that AG2034, an inhibitor of glycinamide ribonucleotide formyltransferase (GARFT) in the pathway for de novo purine synthesis, is cytotoxic to different prostate cancer cell lines in the absence of inosine and hypoxanthine in the culture media. Therefore, we examined DU145 androgen-independent prostate cancer cells selected for drug resistance over a prolonged period of time, in the absence of media components (inosine and hypoxanthine) required for salvage purine synthesis and in the continuous presence of AG2034.
Methods and Results:
DU145 cells were selected over time for 4-[2-(2-amino-4-oxo-4,6,7,8-tetrahydro-3H-pyrimidino[5,4,6][1,4] thiazin-6-yl)-(S)-ethyl]-2,5-thienoylamino-L-glutamic acid (AG2034) resistance by gradually decreasing hypoxanthine concentrations from 1.7 µM to zero in the continuous presence of 10 nM 5-methyl tetrahydrofolate and 50 nM AG2034. Since the inhibition of GARFT blocks glycine incorporation into the purine ring, total cellular ATP was quantified by reverse-phase HPLC and [14C]-glycine incorporation into ATP was determined by liquid scintillation counting of HPLC-isolated ATP fractions. Total and phosphorylated proteins were determined by western blotting. Quantitative RT-PCR was utilized to characterize mRNA expression levels before and after transient transfection studies with small interfering RNA (siRNA). We show that the exposure of parental DU145 cells to AG2034, an inhibitor of GARFT, leads to rapid enzyme inhibition and depletion of ATP. However, resistant DU145 cells are able to re-activate GARFT, restore ATP pools and increase mRNA expression and phosphorylation of ERK1/2 thus, stimulating drug-resistant growth. Moreover, siRNA knockdown of ERK1/2 mRNA in resistant DU145 cells inhibits ERK protein expression and phosphorylation and results in the depletion of ATP and the inhibition of GARFT activity in the absence of changes in GARFT mRNA and protein expression levels.
Conclusions:
The inhibition of ERK1/2 signaling by siRNA transfection affects AG2034 resistance by inhibiting GARFT activity and depleting ATP pools. Hence, we conclude that ERK plays an important role in the maintenance of the AG2034-resistant DU145 phenotype.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 1
First Page: 1
Last Page: 11
Publisher Id: TODRJ-1-1
DOI: 10.2174/1876521101101010001
Article History:
Received Date: 13/10/2010Revision Received Date: 25/2/2011
Acceptance Date: 1/3/2011
Electronic publication date: 28/03/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the University of Medicine and Dentistry of New Jersey, Robert Wood Johnson Medical School, 401 Haddon Avenue, Camden, NJ 08103. USA; Tel: 856-757-2574; Fax: 856-757-2569; E-mail: kemobaj@yahoo.co.uk
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 13-10-2010 |
Original Manuscript | ERK Suppression in DU145 Cells Selected for AG2034 Resistance is Associated with Decreased ATP, GARFT Inactivation and Increased ERK Signaling | |
INTRODUCTION
AG2034 is a rationally designed classical antifolate [1] that is a specific inhibitor of glycinamide ribonucleotide formyltransferase (GARFT), the initial folate requiring enzyme in the de novo purine synthesis pathway. It is transported inside the cell by the reduced folate carrier and is an excellent substrate for polyglutamylation by folylpolyglutamate synthase (FPGS) [1]. This modification aids in drug retention, although the monoglutamate and perhaps the di- and tri-glutamate forms would be considered as substrates for one of the multiple drug resistance protein (MRP) members of the ATP-dependent multidrug efflux transporter family [2]. Indeed, the effectiveness of AG2034 against many different cancer cell lines and xenografts is well-documented [1, 3-5]. However, little is understood regarding the mechanism(s) of resistance to this drug largely because mammalian cells can circumvent the need for GARFT activity for de novo purine synthesis by solely relying on the salvage pathway. To avoid this problem, DU145 prostate cancer cells were selected for drug resistance by culturing cells in the continuous presence of AG2034 in a media that is deficient of inosine and in which hypoxanthine concentrations were periodically lowered to zero. This step-wise process of hypoxanthine reduction gradually prevented the use of the salvage pathway for purine synthesis in these cells. Here-in, we show that in the absence of inosine and hypoxanthine, the primary mechanism of resistance in selected cells is the re-activation of GARFT via markedly overexpressed GARFT mRNA and protein levels, resulting in the restoration of ATP to levels found in drug-naïve parental cells even though growth ensues at a much reduced rate. We also show that resistance is accompanied by an increase in the mRNA expression levels of phosphorylated ERK1/2, and that siRNA knockdown of ERK1/2 in resistant cells results in a rapid decrease in GARFT activity and in the depletion of total ATP. These results therefore implicate ERK in the mechanism of AG2034 resistance.
The extracellular signal-regulated kinase (ERK) cascade (Ras/Raf/MEK/ERK) is a subfamily of the mitogen-activated protein kinase (MAPK) signal transduction pathway and transmits signals from receptors to transcription factors which regulate gene expression, cell cycle progression, proliferation, survival and differentiation of cells [6, 7]. Increased expression and/or activation of the genes in the Ras/Raf/MEK/ERK signaling pathway is often associated with disease progression and with advanced-stage prostate cancer [8-10]. Oncogenic mutations of Ras and/or Raf isoforms also occur in many types of cancer and are major contributors to abnormal cell growth [11-14]. For the treatment of cancer therefore, this pathway has become a therapeutic target because of the central role it plays in the regulation of many cellular processes promoting and/or inhibiting tumor growth and survival. Studies have also shown that the constitutive activation and/or expression of members of the Ras/Raf/MEK/ERK signaling pathway can induce resistance to chemotherapeutic drugs in hematopoietic cells [15].
During its initial stages, prostate cancer is often responsive to androgen ablation therapy [16]. However, it eventually progresses to androgen-independent or hormone-refractory disease [17, 18], which is generally resistant to chemotherapy [19]. Indeed, drug resistance is a major problem that continuously arises during the course of chemotherapy for virtually all cancers and a variety of mechanisms are known to be responsible. These mechanisms include those that affect drug transport, mutations that alter drug targets, and others that modify the agents themselves. The expression of cellular survival pathways in the face of drug challenge can also mediate resistance [20].
MATERIALS AD METHODOLOGY
Chemicals
MEM/F12 medium, trypsin/EDTA, Dulbecco’s Phosphate buffered saline solution (DPBS), penicillin G sodium (10,000 units/ml)/Streptomycin sulfate (10,000 µg/ml) (P/S) and keratinocyte-serum free medium were from Invitrogen/Gibco whereas dialyzed-FBS (d-FBS) was obtained from Valley Biomedical (Winchester, VA). Methyltrienolone, also known as R1881 hormone was purchased from Perkin Elmer. The BCA protein assay kit was obtained from Cole-Parmer Instrument Company (Vernon Hills, IL). [14C (U)]-H2NCH2COOH ([14C]-glycine, 95 mCi/mmol) was obtained from Moravek Biochemicals (Brea, CA). AG2034 was a kind gift from Pfizer (Groton, CT). All other chemicals and reagents were obtained from Sigma-Aldrich (USA), unless otherwise indicated.
Cell Culture
DU145 human prostate cancer cells were purchased from the American Type Culture Collection (Bethesda, MD). Cells were initially cultured in DMEM/F12 medium supplemented with 10% FBS and penicillin/streptomycin (100units/ml, 100 µg/ml, respectively). Cells were then maintained in FA-Plus medium [21, 22] before all experimental procedures. FA-Plus medium contains MEM/F12 (deficient in hypoxanthine, inosine, thymidine, folate, L-glutamine and phenol red) supplemented with 10% dialyzed-FBS, 1.7 µM hypoxanthine, 1.5 µM thymidine, 0.05 nM R1881 hormone, 10 nM 5-methyl tetrahydrofolate, penicillin (100 units/ml)/streptomycin (100 µg/ml) and 2 mM L-glutamine. Cells were cultured as monolayers in a humidified environment of 5% CO2/95% air at 37ºC.
Cell Selection for Resistance to AG2034
Cells (5 x 105) in FA-Plus medium were plated in 100-mm petri dishes and allowed 48 h to adhere to plates. Culture medium was renewed every 2-3 days with FA-Plus containing 50 nM AG2034 for 14 days. Thereafter, cell monolayers were washed with DPBS and the media was replaced with FA-Plus medium containing 0.85 µM hypoxanthine and 50 nM AG2034 for 7 days. Cells from 5-petri dishes were harvested, pooled and re-plated at 5 x 105 in FA-Plus containing 0.425 µM hypoxanthine and 50 nM AG2034 for another 7 days. The media was then renewed with FA-Plus containing 0.106 µM hypoxanthine and 50 nM AG2034. After 14 days, cells were trypsinized and re-plated at 1 x 104 per well in 6-well plates in FA-Plus containing 0.106 µM hypoxanthine and 50 nM AG2034 and allowed 48 h to adhere. Finally, cell monolayers were washed with DPBS and media was replaced with FA-Plus without hypoxanthine (0HPX media) again containing 50 nM AG2034 with media renewal every 2-3 days. Cells were trypsinized and re-plated every 2 weeks in 0HPX media containing 50 nM AG2034. After 6 months, a sub-population of cells initiated growth, albeit slowly, thus displaying resistance to the continuous presence of AG2034.
Cell Cloning
Resistant cells were harvested and seeded at 50 cells into 100-mm petri dishes in order to obtain colonies that were sufficiently separated to allow for the isolation of individual colonies. These cells were maintained in 0HPX medium containing 50 nM AG2034. After 48 h, each dish was microscopically observed and dishes containing adhered well spaced single cells were scored. Media was renewed every 2-3 days. After 6 weeks, cell monolayers were washed with DPBS, sterile cloning rings were dipped into sterilized stopcock grease and placed firmly against the plastic plates upon which growing cell colonies were previously scored. Colonies were separately trypsinized and each transferred into separate wells of 6-well plates containing fresh 0HPX media supplemented with 50 nM AG2034.
Cell Growth Assay
Parental DU145 cells or cloned resistant DU145 cells were plated at 1 x 103 per well in 6-well plates. Cells were then maintained in 0HPX media with or without 50 nM AG2034 for varying periods of time. Cells were harvested by trypsinization and aliquots counted electronically using a Beckman Coulter Counter. Culture medium was renewed with freshly-prepared medium every 2-3 days for 13 days. Images of the cells were taken on an inverted phase-contrast microscope (32X magnification) on day-12 of the experiment.
Cell Treatments Prior to Nucleotide Extraction and [14C]-Glycine Incorporation Assay
In the first set of experiments, cells (1 x 106) were allowed 48 h to adhere to plates in 0HPX media with or without 50 nM AG2034. Thereafter, 0.8 µCi/ml of [14C]-glycine was added for a further 24 h. As a positive control, drug naïve parental DU145 cells were treated with 50 nM AG2034 for the final 24 h of the experiment. In other experiments, following siRNA transfections (see below), cells from 24-wells were pooled together to obtain sufficient numbers for nucleotide detection and [14C]-glycine incorporation studies on the HPLC system.
Nucleotide Extraction and Analysis
Cell monolayers were washed twice with ice-cold DPBS, harvested by trypsinization and lysed by the addition of trichloroacetic acid (0.73 M, final concentration). The resulting suspension was cleared by centrifugation and an aliquot of the supernatant was neutralized with ice-cold KOH. Total cellular ATP was quantified by reverse-phase HPLC of trichloroacetic acid (TCA) extracts using a Vydac C18, reverse-phase column, as previously described [21, 22]. Protein concentration was determined spectrophotometrically using the BCA protein assay kit.
[14C]-Glycine Incorporation Assay
[14C]-glycine incorporation into ATP was determined by collecting ATP fractions after nucleotide separation by HPLC as previously described [21]. An aliquot of the ATP fraction was assayed for radioactivity by liquid scintillation counting. [14C]-glycine incorporation into ATP is expressed as a percentage of the total incorporated radioactivity into the ATP fractions of the drug naïve parental cells.
Human Phospho-MAPK Profile Array Assay
Cells (parental and drug-resistant) were rinsed with DPBS, lysed according to the manufacturer’s instructions using the human phospho-MAPK array kit (R&D systems, Minneapolis, MN). Protein concentration of lysates was determined spectrophotometrically using the BCA protein assay kit. The arrays were incubated overnight with 1.2 mg of cell lysates at 2-8ºC, washed and detected using the manufacturer’s provided detection antibody cocktail. Arrays were then developed using the HyGLO chemiluminescent detection reagent (Denville scientific, Metuchen, NJ).
Western Blotting
Cells were lysed in ice-cold RIPA buffer (Phosphate buffered saline, 3.5 mM sodium dodecyl sulfate, 11.6 mM sodium deoxycholate, 16.3 mM Nonidet P-40) supplemented with 1 mM Na3VO4, 0.57 mM phenylmethylsulfonyl fluoride, and 7.7 µM aprotinin. Protein concentrations were determined with the BCA assay (Pierce, Rockford, IL) using bovine serum albumin as the standard. Equal amounts of protein were mixed with SDS loading buffer, boiled and separated on Tris-HCl gels. Proteins were then transferred onto PVDF membranes (Bio-rad, Hercules, CA) and blocked with 5% non-fat dry milk. Membranes were probed with specific antibodies against ERK1/2, phospho-ERK1/2 (Thr 202/Tyr 204), β-actin (Cell signaling technology, Beverly, MA) and GARFT (Santa Cruz Biotechnology, CA). The appropriate horseradish peroxidase-conjugated secondary antibodies were used and developed using the HyGLO chemiluminescent detection reagent (Denville scientific, Metuchen, NJ).
Reverse Transcription and Real Time PCR
Total RNA was isolated from cells using the PerfectPure RNA cultured cell kit (5 Prime Inc.). cDNA was synthesized from 3 µg of total RNA in a final volume of 50 µl using the high capacity cDNA reverse transcription kit (Applied Biosystems). Thermal cycling conditions included 10 minutes at 25°C, 2 h at 37°C, 5 minutes at 85ºC and then held at 4ºC for <12 h.
Real Time PCR reactions were performed in a total volume of 20 µl using the Taqman Master Mix reagent (Applied Biosystems) in the 7900 Fast Real Time PCR instrument (Applied Biosystems). Reactions included primers and FAM dye-labeled probe sets specifically targeting ERK1, ERK2, MEK1, MEK2, B-Raf, N-Ras, GARFT and GAPDH, the housekeeping gene. Thermal cycling conditions included 2 minutes at 50°C and 10 minutes at 95°C. This was followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Threshold cycle (CT) values greater than 40 indicate absence of the transcript of interest. Results were analyzed using the sequence detection system (SDS) version 2.2.2 (Applied Biosystems), normalized to controls and expressed as relative fold-changes in gene expression using the 2-∆∆CT method. All primers used were validated RealTime PCR primers purchased from Applied Biosystems.
siRNA
siRNA probes targeting ERK1 &2 expression were purchased from Ambion (Austin, TX). Cells were plated at 1 x104 cells/well, in 24-well plates, and allowed ~24 h to adhere. Cells were then transiently transfected with specific ERK1&2 probes (Ambion) or non-targeting DNA control probes (Ambion) using the siPORT lipid transfection reagent (Ambion) according to manufacturer’s protocol. Briefly, siPORT lipid transfection reagent was incubated with Opti-MEM reduced serum medium (Invitrogen) for 10 minutes at room temperature. Specific ERK1&2 probes or non-targeting DNA probes were added to siPORT lipid mixture and incubated at room temperature for 15 min for proper transfection complex formation. Transfection mixtures were then added to cells in serum-free media. After 4 h of incubation at 37ºC/5% CO2, 1ml of 0HPX media with (resistant cells) or without (parent cells) 50 nM AG2034 (final concentration) was added to each well. In cells utilized for ATP and [14C]-glycine incorporation experiments, the 1 ml of added media contained 0.63 µCi/ml of [14C]-glycine. After a further 30 h of transfection, RNA and protein were isolated from cells. Cellular nucleotides were also extracted for total ATP determinations and [14C]-glycine incorporation.
Statistical Analysis
Data shown are from three independent experiments. Comparisons were made by one-way analysis of variance (ANOVA) or student’s t-test using MINITAB statistical software, version 13.32. Post hoc comparisons of means were made using Tukey’s pairwise method. Differences were considered significant at P < 0.05.
RESULTS
AG2034-Resistant DU145 Cells Proliferate at a Much Slower Rate Than Drug-Naïve Parental Cells
Measurement of cell proliferation over 13 days in the hypoxanthine deficient media (0HPX media) allowed for the determination of the differences in growth rate between parental and cloned drug-resistant cells in the presence or absence of AG2034 (Fig. 1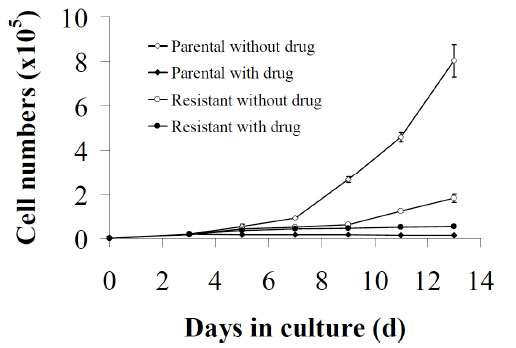 ). There was no significant difference in cell numbers between drug-untreated or drug-treated parental cells and resistant cells after 3 days of plating cells with or without AG2034. On day 5, parental cells, as well as resistant cells, either in the absence or presence of 50 nM AG2034, proliferated at the same rate. By day 7 however, the number of cells in the non-treated parental cell population was 6-fold higher than that in the treated parental cell population and 2.2-fold more than the resistant cell population even though the same number of cells had been initially plated. On days 11 and 13, the number of non-treated parental cells was higher by ~31-fold and ~77-fold, respectively, in comparison to the drug-treated parental population but only ~8 and ~12-fold greater than the drug-treated resistant cell population. Indeed, by day 13 the number of cells in the drug-treated resistant cell population was 6.6-fold higher than that in the drug-treated parental population. Importantly, the doubling time for drug-naïve parental cells in 0HPX media is 25.1 hours while that for the resistant cells when continuously maintained in the presence of drug is 9.7 days. When the parental cells are treated with 50 nM AG2034, however, the cells stop growing and over 35 days of monitoring growth [21], these cells never resume proliferation. Interestingly, when the drug is withdrawn from the culture media in which the resistant cells are maintained, the doubling time reduces to 6.8 days. While these results (Fig. 1) confirm that in the continuous presence of AG2034, parental DU145 cells cease to grow [21] and show that cells selected for resistance under these conditions are able to re-initiate growth although at a very slow rate, the increase in growth rate of the resistant cells post AG2034 withdrawal suggests that continuous drug presence may have secondary effects on growth.
). There was no significant difference in cell numbers between drug-untreated or drug-treated parental cells and resistant cells after 3 days of plating cells with or without AG2034. On day 5, parental cells, as well as resistant cells, either in the absence or presence of 50 nM AG2034, proliferated at the same rate. By day 7 however, the number of cells in the non-treated parental cell population was 6-fold higher than that in the treated parental cell population and 2.2-fold more than the resistant cell population even though the same number of cells had been initially plated. On days 11 and 13, the number of non-treated parental cells was higher by ~31-fold and ~77-fold, respectively, in comparison to the drug-treated parental population but only ~8 and ~12-fold greater than the drug-treated resistant cell population. Indeed, by day 13 the number of cells in the drug-treated resistant cell population was 6.6-fold higher than that in the drug-treated parental population. Importantly, the doubling time for drug-naïve parental cells in 0HPX media is 25.1 hours while that for the resistant cells when continuously maintained in the presence of drug is 9.7 days. When the parental cells are treated with 50 nM AG2034, however, the cells stop growing and over 35 days of monitoring growth [21], these cells never resume proliferation. Interestingly, when the drug is withdrawn from the culture media in which the resistant cells are maintained, the doubling time reduces to 6.8 days. While these results (Fig. 1) confirm that in the continuous presence of AG2034, parental DU145 cells cease to grow [21] and show that cells selected for resistance under these conditions are able to re-initiate growth although at a very slow rate, the increase in growth rate of the resistant cells post AG2034 withdrawal suggests that continuous drug presence may have secondary effects on growth.

A series of photomicrographs, (Fig. 2 ), showed that drug-treated parental cells were elongated after 24 h of treatment and that this persisted for the 12-day duration of the experiment (Fig. 2B). As observed previously [21], some of these cells became detached from the plate indicating that in the absence of hypoxanthine the drug was cytotoxic. However, untreated parental cells remained adhered to the plate maintaining a flattened morphology (Fig. 2A) characteristic of the cell type under these growth conditions where the cells rely on the de novo pathway for purine synthesis in the absence of drug and hypoxanthine. These characteristics were also found to be present in drug-resistant cells (Fig. 2C) that were able to proliferate in the presence or absence of drug (Fig. 1). Hence, the acquisition of resistance to AG2034 had been accompanied both by a return to a more adherent and flattened morphology and the ability to re-initiate proliferation.
), showed that drug-treated parental cells were elongated after 24 h of treatment and that this persisted for the 12-day duration of the experiment (Fig. 2B). As observed previously [21], some of these cells became detached from the plate indicating that in the absence of hypoxanthine the drug was cytotoxic. However, untreated parental cells remained adhered to the plate maintaining a flattened morphology (Fig. 2A) characteristic of the cell type under these growth conditions where the cells rely on the de novo pathway for purine synthesis in the absence of drug and hypoxanthine. These characteristics were also found to be present in drug-resistant cells (Fig. 2C) that were able to proliferate in the presence or absence of drug (Fig. 1). Hence, the acquisition of resistance to AG2034 had been accompanied both by a return to a more adherent and flattened morphology and the ability to re-initiate proliferation.

The Comparison of Cellular ATP Levels, [14C]-Glycine Incorporation, GARFT mRNA and Protein Levels Between Parental and Resistant DU145 Cells
In order to determine whether the difference in growth rate between resistant and drug-naïve parental cells is associated with differences in total cellular ATP, levels of this nucleotide was determined by reverse-phase HPLC. To this end, parental DU145 cells were treated with 50 nM AG2034 for 24 h and total cellular ATP was quantified and compared to resistant cells maintained in the continuous presence of AG2034. Fig. (3A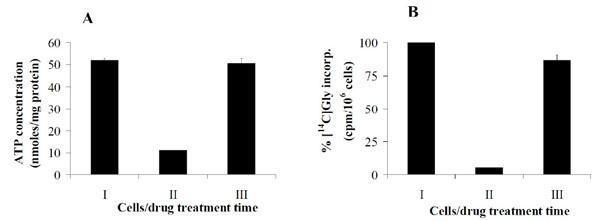 ) shows that the treatment of parental cells with 50 nM AG2034 for 24 h reduced the steady state level of ATP from approximately 52 to 11 nmoles/mg protein (Fig. 3AI, AII). This demonstrates the effectiveness of the drug at depleting total ATP levels. As previously reported [21], ATP levels in parental DU145 cells exposed to 50 nM AG2034 and cultured in the absence of hypoxanthine remain significantly depleted for 35 days. Interestingly, there was no significant difference in ATP pool size between parental cells (Fig. 3AI) and resistant cells (Fig. 3AIII), even though their growth rate was substantially different (Fig. 1). Hence, resistant cells had restored their ATP pool levels independent of their inability to proliferate at parental control levels. These results show that in resistant cells growth rate is independent of ATP pool size. They also show that under conditions where AG2034 is present and salvage purine synthesis is prevented by the absence of hypoxanthine and inosine in the media, ATP pool size can be restored.
) shows that the treatment of parental cells with 50 nM AG2034 for 24 h reduced the steady state level of ATP from approximately 52 to 11 nmoles/mg protein (Fig. 3AI, AII). This demonstrates the effectiveness of the drug at depleting total ATP levels. As previously reported [21], ATP levels in parental DU145 cells exposed to 50 nM AG2034 and cultured in the absence of hypoxanthine remain significantly depleted for 35 days. Interestingly, there was no significant difference in ATP pool size between parental cells (Fig. 3AI) and resistant cells (Fig. 3AIII), even though their growth rate was substantially different (Fig. 1). Hence, resistant cells had restored their ATP pool levels independent of their inability to proliferate at parental control levels. These results show that in resistant cells growth rate is independent of ATP pool size. They also show that under conditions where AG2034 is present and salvage purine synthesis is prevented by the absence of hypoxanthine and inosine in the media, ATP pool size can be restored.

Although the recycling of purines via RNA degradation could contribute to ATP pool size, it is unlikely that its contribution would be sufficient to restore sizes to parental cell levels even though the growth rate of resistant cells is very slow whether the drug is continuously present or withdrawn (Fig. 1). Therefore, we considered the possibility that the de novo pathway had been re-activated. As shown in Fig. (3B), GARFT activity had been restored in resistant cells (Fig. 3BIII) as indicated by their ability to incorporate [14C]-glycine into ATP to near untreated parental cell levels (Fig. 3BI), albeit in the presence of 50 nM AG2034. As previously reported this drug concentration is sufficient to inhibit GARFT activity by >90% after 24 h of drug exposure (Fig. 3BII), regardless of the presence or absence of hypoxanthine in the media [21]. The results in Fig. (4A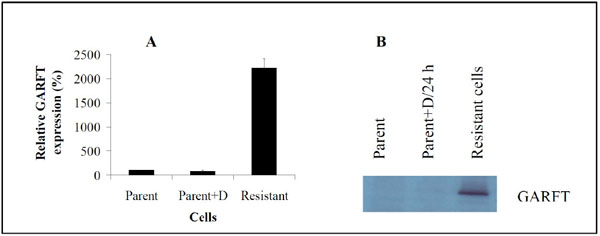 , B) indicate that the increase in GARFT activity in the resistant cells is mirrored both by a dramatic increase in GARFT mRNA and protein expression levels, respectively, largely explaining the ability of these cells to restore their ATP pools (Fig. 3A).
, B) indicate that the increase in GARFT activity in the resistant cells is mirrored both by a dramatic increase in GARFT mRNA and protein expression levels, respectively, largely explaining the ability of these cells to restore their ATP pools (Fig. 3A).

Phosphorylated ERK is Upregulated in AG2034-Resistant DU145 Prostate Cancer Cells
Since the Ras/Raf/MEK/ERK cascade of the MAPK signal transduction pathway plays a pivotal role in the regulation of cancer cell growth and survival [7], we sought to determine whether this pathway was upregulated in DU145 cells selected for resistance to AG2034. Firstly, total proteins were isolated from drug-naïve parental and drug-resistant cells utilizing the lysis buffer provided in the human phospho-MAPK profile array kit. As shown in Fig. (5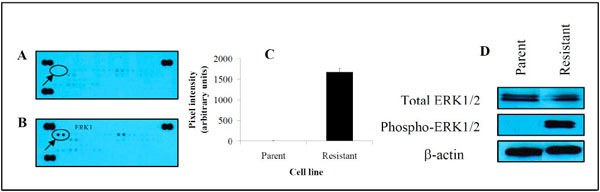 ), drug resistance induced a marked increase in the phosphorylation status of ERK1 by ~480-fold (Fig. 5B, 5C) in comparison to parental DU145 cells (Fig. 5A, 5C) when cultured in the absence of hypoxanthine (0HPX media).
), drug resistance induced a marked increase in the phosphorylation status of ERK1 by ~480-fold (Fig. 5B, 5C) in comparison to parental DU145 cells (Fig. 5A, 5C) when cultured in the absence of hypoxanthine (0HPX media).

To confirm the level of ERK expression in resistant cells, immunoblot assays of total and phosphorylated forms of ERK1 and 2 were performed. There was no difference in total ERK1/2 protein levels between parental and resistant DU145 cells (Fig. 5D). In support of the results obtained in Fig. (5C), phosphorylated ERK1/2 was clearly present in resistant cells but undetectable in parental cells. These results suggest that the activation of ERK1/2 in DU145 cells is associated with the acquisition of resistance to AG2034 and plays a significant role in the survival mechanism employed by these cells in response to drug challenge.
To determine if the increase in ERK1/2 protein expression and phosphorylation levels in resistant cells was commensurate with increased ERK1/2 mRNA expression levels, we utilized quantitative RT-PCR. Fig. (6A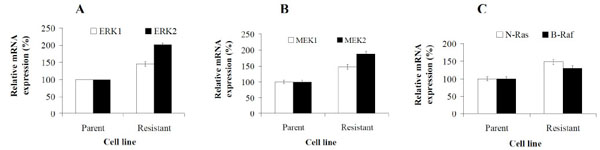 ) shows that resistant cells increase their mRNA expression levels of both ERK1 and ERK2 by ~1.5- and 2-fold, respectively, in comparison to parental cells. To evaluate whether the activation of ERK in resistant cells is associated with similar increases in its upstream activators, MEK1/2, B-Raf and N-Ras, we also compared the expression levels of these genes in parental and resistant cells. Analysis revealed that in resistant cells the expression levels of MEK1 and 2 increased by ~1.5 and 2-fold, respectively when compared to parental cells (Fig. 6B). In addition, resistant cells also exhibited significantly increased mRNA expression levels of N-Ras and B-Raf (Fig. 6C) by 1.3 and ~1.5-fold, respectively, versus parental cell levels. These results show that in AG2034 resistant cells, activation of ERK1/2 signaling is associated with increased expression of its upstream regulators. Hence, the ERK signaling pathway is likely to play an important role in conferring resistance thus, contributing to the survival of prostate cancer cells under conditions of drug challenge.
) shows that resistant cells increase their mRNA expression levels of both ERK1 and ERK2 by ~1.5- and 2-fold, respectively, in comparison to parental cells. To evaluate whether the activation of ERK in resistant cells is associated with similar increases in its upstream activators, MEK1/2, B-Raf and N-Ras, we also compared the expression levels of these genes in parental and resistant cells. Analysis revealed that in resistant cells the expression levels of MEK1 and 2 increased by ~1.5 and 2-fold, respectively when compared to parental cells (Fig. 6B). In addition, resistant cells also exhibited significantly increased mRNA expression levels of N-Ras and B-Raf (Fig. 6C) by 1.3 and ~1.5-fold, respectively, versus parental cell levels. These results show that in AG2034 resistant cells, activation of ERK1/2 signaling is associated with increased expression of its upstream regulators. Hence, the ERK signaling pathway is likely to play an important role in conferring resistance thus, contributing to the survival of prostate cancer cells under conditions of drug challenge.

Inhibition of ERK1/2 Expression by siRNA in Drug- Resistant Cells is Associated with the Depletion of ATP Pools and Deactivation of GARFT Activity without Affecting GARFT mRNA or Protein Levels
Since the acquisition of resistance to AG2034 by DU145 cells required ~6 months of selection to yield cells characterized by an upregulated Ras/Raf/MEK/ERK signaling pathway, a restored ATP pool size to normal parental levels and the reactivation of GARFT, we asked if the inhibition of ERK1/2 in resistant cells would have a direct effect on ATP and GARFT activity. Notably, the pretreatment of these resistant DU145 clones for up to 7 days with up to 50 µM levels of ERK inhibitors, PD98059 [23] and U0126, had no effect on cell proliferation but rather elevated the mRNA expression levels of both ERK 1 and 2 (data not shown). Therefore, both parental and resistant cells were transfected with siRNA probes specifically targeting ERK1/2 mRNA. Effective transient knockdown of ERK mRNA expression was observed in both cell lines, producing approximately 86% reduction in ERK1/2 mRNA in parental cells (Fig. 7A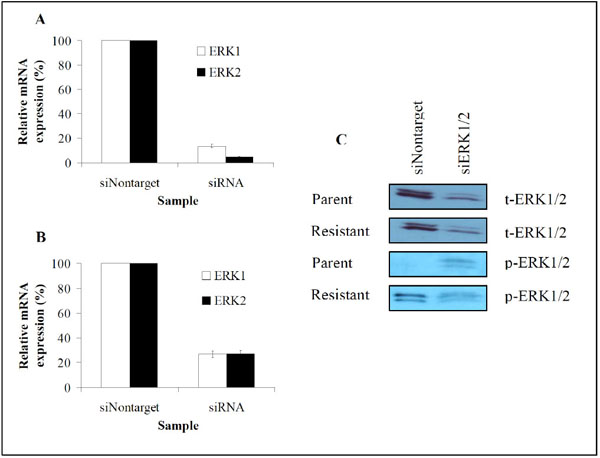 ) and 73% reduction in resistant cells (Fig. 7B). Immunoblot assays further showed that reductions in mRNA levels were accompanied by similar decreases in total and phosphorylated ERK1/2 protein levels (Fig. 7C) in the resistant cells. However in the parental cells, ERK phosphorylation was increased (Fig. 7C).
) and 73% reduction in resistant cells (Fig. 7B). Immunoblot assays further showed that reductions in mRNA levels were accompanied by similar decreases in total and phosphorylated ERK1/2 protein levels (Fig. 7C) in the resistant cells. However in the parental cells, ERK phosphorylation was increased (Fig. 7C).

As shown in Fig. (8A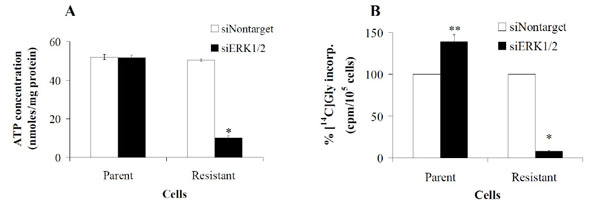 ), a 34 h transient transfection of ERK 1 and 2 specific siRNAs into parental DU145 cells had no effect on ATP pools nor cell morphology (data not shown). In contrast however, ATP pool size in resistant cells reduced from 52 to 10.2 nmoles/mg protein. Moreover, [14C]-glycine incorporation into ATP in resistant cells was inhibited by 92% from 3093 cpm/105 cells to 254 cpm/105 cells (Fig. 8B), directly correlating the decrease in ATP pool size with the loss of GARFT activity. ERK inhibition with siRNA post 34 hours of transfection had no effect on the morphology of parental and resistant cells (data not shown).Cell proliferation in parental and resistant cells was static for the 34 h duration of transient transfection with siERK1/2 but growth ensued thereafter (data not shown). Surprisingly, the loss of activity in the resistant cells was not accompanied by decreases in GARFT mRNA (Fig. 9A
), a 34 h transient transfection of ERK 1 and 2 specific siRNAs into parental DU145 cells had no effect on ATP pools nor cell morphology (data not shown). In contrast however, ATP pool size in resistant cells reduced from 52 to 10.2 nmoles/mg protein. Moreover, [14C]-glycine incorporation into ATP in resistant cells was inhibited by 92% from 3093 cpm/105 cells to 254 cpm/105 cells (Fig. 8B), directly correlating the decrease in ATP pool size with the loss of GARFT activity. ERK inhibition with siRNA post 34 hours of transfection had no effect on the morphology of parental and resistant cells (data not shown).Cell proliferation in parental and resistant cells was static for the 34 h duration of transient transfection with siERK1/2 but growth ensued thereafter (data not shown). Surprisingly, the loss of activity in the resistant cells was not accompanied by decreases in GARFT mRNA (Fig. 9A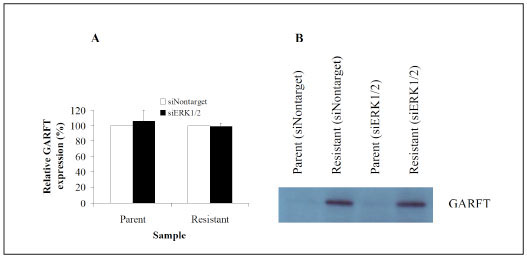 ) or protein (Fig. 9B) levels. Interestingly, similar transfection of these siRNA probes into parental control cells actually elevated the incorporation of [14C]-glycine into ATP by 39% as compared to non-target siRNA control probes. Overall these results imply that in AG2034-resistant DU145 cells, the sustained activation of ERK is required to maintain ATP pool size and GARFT activity.
) or protein (Fig. 9B) levels. Interestingly, similar transfection of these siRNA probes into parental control cells actually elevated the incorporation of [14C]-glycine into ATP by 39% as compared to non-target siRNA control probes. Overall these results imply that in AG2034-resistant DU145 cells, the sustained activation of ERK is required to maintain ATP pool size and GARFT activity.


DISCUSSION AND CONCLUSION
Previously, we showed that AG2034 is an excellent inhibitor of proliferation for non-tumorigenic prostate epithelial cells [22] as well as androgen-dependent [22] and androgen-independent [21] prostate cancer cells. In the presence or absence of near physiological levels of hypoxanthine, 1.7 µM, the dose of AG2034 required to inhibit the growth of the prostate cancer cell line, DU145, to 50% of controls after 5 d of drug treatment (ED50) was 4 nM and increasing the dose to 40 nM did not significantly further decrease cell numbers nor did it permit any growth during the 120 h assay. Hence, under these conditions for 5 days, AG2034 acted as a cytostatic agent. Clonogenic assays showed that AG2034 was actually cytotoxic to DU145 cells in the absence of hypoxanthine, with an LD50 of 50 nM, but even at that elevated dose, some colonies remained on the plate for the 14 d time frame of the experiment [21]. The data in Figs. (1, 2) are consistent with these results showing that in the continuous presence of 50 nM AG2034, DU145 cells cultured in the absence of hypoxanthine do not grow but become elongated and most of the cells lose their ability to remain adhered to the plate. Selection for resistance therefore relied upon the need for these cells, in the presence 50 nM AG2034, to maintain their ability to synthesize purines via salvage as the concentration of hypoxanthine was continuously decreased to zero over a short period of time (~30 days). This process of decreasing hypoxanthine levels in a step-wise manner over ~30 days resulted in the selection of slow growing, AG2034-resistant clones (Fig. 1).
The effect of AG2034 on parental DU145 cells was as previously reported [21] in that GARFT activity was severely inhibited (Fig. 3BI, II) and ATP pools were depleted (Fig. 3AI, 3II). Restoration of ATP pool size and GARFT activity in the slow-growing resistant cell clones (Fig. 1) demonstrate that the mechanism of resistance clearly includes the reactivation of GARFT and the subsequent restoration of the de novo purine synthesis pathway since salvage in the absence of inosine and hypoxanthine in the media cannot explain restoration of ATP pool size. RNA turnover in the absence of growth, and hence DNA synthesis, could account for the prolonged (~6 months) cytostasis of these cells allowing for some level of purine recovery and re-use. Nevertheless, it is highly unlikely that such a turnover would be sufficient to restore ATP pools to parental cell levels as observed in the slow-growing resistant cells, and at the same time support growth, even at such low rates. In resistant cells, restoration of ATP levels (Fig. 3A) and GARFT activity (Fig. 3B) was accompanied by increases in expression levels of GARFT mRNA (Fig. 4A) and protein (Fig. 4B) demonstrating that the mechanism of resistance includes the reactivation of GARFT and subsequent restoration of the de novo purine synthesis pathway. That this occurs in the presence of 50 nM AG2034 suggests that the level of GARFT expression in resistant cells is sufficient to overcome the effects of the drug. Indeed, results in Fig. (4A) show that the amount of GARFT mRNA in resistant cells is dramatically upregulated by ~2200% while the amount of GARFT protein although hardly detectable in parental cells, is substantially increased. Since AG2034 is a classical antifolate, reversal of its activity is subject to a variety of mechanisms [24] including the inactivation of FPGS, mutations in the drug target protein (GARFT) that could alter the drug binding site thereby rendering the drug ineffective and elevations in the expression of MRP transport proteins [25]. While these may play a role in the overall mechanism of resistance, we conclude that the reactivation of GARFT via its overexpression plays a major role in the development of the resistance phenotype and is responsible for restoration of ATP pool size. That the decrease in GARFT activity and ATP pool size in resistant cells after knockdown of ERK1/2 expression is not accompanied by concurrent decreases in GARFT mRNA and protein levels was unexpected and remains unexplained. One possibility is that ERK1/2 may affect GARFT mRNA splicing such that the resulting mRNA and protein produced are similar in size to that found to be overexpressed in resistant cells, but are nonfunctional. Further studies will be required to explain this result.
The components of the Ras/Raf/MEK/ERK signaling pathway are often altered in tumor cells and conflicting evidence exists regarding its contribution to the development of resistance to several chemotherapeutic agents. Also, the role of this pathway in the development of drug resistance is unclear. The results of the phospho-MAPK profile array and western blotting experiments represented in Fig. (5) indicate that phosphorylated ERKs are overexpressed in resistant DU145 cells as are the RNA expression levels of MEK1 and 2, N-Ras, and B-Raf (Fig. 6). These results clearly suggest that the ERK signaling pathway is involved in the acquisition of resistance to AG2034 by DU145 cells. This finding is consistent with an increasing body of evidence indicating that altered gene expression or activity within the Ras/Raf/MEK/ERK pathway, known to be responsible for regulating many cellular processes including proliferation, growth and differentiation [26], may increase the risk of drug resistance in different cancer cell types [27-30], thus underscoring its potential relevance to the all too frequent clinical failure of chemotherapy [7, 29, 31]. In other studies using advanced prostate cancer cells in which the expression of Raf, MEK, and ERK genes is low, it has been reported that while the Raf/MEK/ERK pathway may be induced by treatment with paclitaxel or doxorubicin, resistance to these drugs is independent of Raf/MEK/ERK signaling [32]. In contrast, it has also been reported that the activation of this pathway is important only to inhibit proliferation and induce cancer cell death [33-35]. As far as resistance to antifolates is concerned, little is known about the role played, if any, by the ERK signaling pathway. It has been reported that activation of the pathway can alter the expression of ATP dependent drug efflux pumps and promote anti-apoptotic responses which contribute to the development of resistance to chemotherapeutic drugs [7]. Under the conditions studied here, the enhancement of the expression of anti-apoptotic and other pro-survival proteins is likely, as indicated by the fact that the selection process yields viable, though, slow growing cells (Fig. 1) while continuously exposed to a cytotoxic agent. We have reported that AMP-activated protein kinase (AMPK) is activated via phosphorylation on Thr-172, in DU145 cells within 24 h of exposure to AG2034 [21]. Since it has been shown that AMPK is a key activator of ERK and that the cross talk between these two pathways couples cell survival to energy status [36], it is reasonable to suggest that in these resistant cells, the function of ERK is to support cell survival rather than to promote apoptosis.
Although ERK1 & 2 are the only known substrates for MEK1/2, there may be yet unknown substrates for this gene. Nonetheless, the MEK/ERK pathway regulates gene expression and promotes tumor growth and survival [7, 37]. Here-in, we show that the upregulation of the ERK genes in AG2034-resistant clones correlates with increased basal expression levels of the upstream regulatory genes, N-Ras, B-Raf and MEK (Fig. 6). Early reports suggested that different cells respond differently to Raf expression depending on the isoform that is overexpressed [38, 39]. In NIH-3T3 fibroblasts and FDC-P1 hematopoietic cells for instance, the overexpression of B-Raf led to cell growth arrest whereas A-Raf or Raf-1 overexpression caused the cells to proliferate [38-41]. Although there was no significant difference in mRNA expression levels of A-Raf or Raf-1 in the androgen-independent prostate cancer DU145 resistant clones used in this study (data not shown), upregulation of the B-Raf isoform (Fig. 6C ), neither induced cell growth arrest (Fig. 1) nor affected cell cycle progression (data not shown). However, this may be because it was only moderately upregulated [38].
), neither induced cell growth arrest (Fig. 1) nor affected cell cycle progression (data not shown). However, this may be because it was only moderately upregulated [38].
The results in Fig. (7) show that transient transfection of ERK1/2 specific siRNA probes into untreated and drug-resistant DU145 cells effectively decreased the expression of ERK1/2 in both cell types. As a result, activated ERK1/2 is downregulated in resistant cells. Unexplained is the observation that in transfected non-drug-treated parental cells, phosphorylated ERK is present (Fig. 7C), albeit at very low levels, whereas it is not detected in non-transfected parental cells (Fig. 5). It may be possible that the transfection process itself, possibly due to stress, induced low level expression and activation of ERK. This, however, remains to be clarified. While the transient transfection of ERK specific siRNA probes increased phosphorylation of ERK1/2 in the parental cell line (Fig. 7C), it had no effect on total ATP levels (Fig. 8A), but correlates with an increase in GARFT activity as measured by [14C]-glycine incorporation into ATP (Fig. 8B). Interestingly, the opposite effect occurred in resistant cells where knockdown of ERK1/2 was correlated with a dramatic loss of ATP (Fig. 8A) and GARFT activity (Fig. 8B), indicating that ERK expression has a direct effect on GARFT activity in the resistant cells. The lack of an effect on ATP pool size in parental cells (Fig. 8B) is not surprising given how such pools are maintained under normal growth conditions. However, the elevation in GARFT activity (Fig. 8B) in parental cells was unexpected. While the data in Fig. (9) indicate that the knockdown of ERK has no effect on GARFT mRNA (Fig. 9A) or protein expression (Fig. 9B) levels, the results in Fig. (8B) show that the knockdown of ERK inhibits GARFT activity in the resistant clones. Taken together, these results suggest that ERK may play different roles under normal as opposed to resistant cell conditions in the regulation of GARFT activity.
ACKNOWLEDGEMENTS
This work was supported by the State of New Jersey and by the Robert Wood Johnson School of Medicine. We thank Pfizer (Groton, CT) for the supply of AG2034 used in this study.