- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Drug Discovery Toward Antagonists of Methyl-Lysine Binding Proteins
J. Martin Herold, Lindsey A Ingerman, Cen Gao, Stephen V Frye*
Abstract
The recognition of methyl-lysine and -arginine residues on both histone and other proteins by specific “reader” elements is important for chromatin regulation, gene expression, and control of cell-cycle progression. Recently the crucial role of these reader proteins in cancer development and dedifferentiation has emerged, owing to the increased interest among the scientific community. The methyl-lysine and -arginine readers are a large and very diverse set of effector proteins and targeting them with small molecule probes in drug discovery will inevitably require a detailed understanding of their structural biology and mechanism of binding. In the following review, the critical elements of methyl-lysine and -arginine recognition will be summarized with respect to each protein family and initial results in assay development, probe design, and drug discovery will be highlighted.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 5
Issue: Suppl 1
First Page: 51
Last Page: 61
Publisher Id: CCGTM-5-51
DOI: 10.2174/1875397301005010051
Article History:
Received Date: 26/1/2011Revision Received Date: 22/2/2011
Acceptance Date: 25/4/2011
Electronic publication date: 22/8/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Center for Integrated Chemical Biology and Drug Discovery, UNC Eshelman School of Pharmacy, Division of Medicinal Chemistry and Natural Products, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA; Tel: 919-843-5486; Fax: 919-843-8465; E-mail: svfrye@email.unc.edu
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 26-1-2011 |
Original Manuscript | Drug Discovery Toward Antagonists of Methyl-Lysine Binding Proteins | |
INTRODUCTION AND PROTEIN FAMILIES
Methylation of lysine and arginine residues on histones and other proteins plays a crucial role in the activation and repression of gene expression, and consequently, the study of epigenetic regulation has recently emerged as both a major challenge and an opportunity in biomedical sciences [1Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis Nat Struct Mol Biol 2007; 14: 1008-6., 2Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, Part I: Covalent histone modifications Trends Mol Med 2007; 13: 363-72.]. Methylation is often considered to be one letter in a diverse alphabet of histone post-translational modifications (PTMs), including phosphorylation, ubiquination, acetylation, and glycosylation, among others [3Jenuwein T, Allis CD. Translating the histone code Science 2001; 293: 1074-80., 4Strahl BD, Allis CD. The language of covalent histone modifications Nature 2000; 403: 41-5.]. PTMs play a critical role in the regulation of signaling pathways where they can serve as chemical switches to induce or repress protein-protein interactions [5Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails Genes Dev 2001; 15: 2343-60.]. In particular, methylation marks have been shown to serve as initiators for the recruitment of non-histone proteins which dictate higher-order chromatin structure and function, such as gene expression and repression [3Jenuwein T, Allis CD. Translating the histone code Science 2001; 293: 1074-80., 6Kouzarides T. Chromatin modifications and their function Cell 2007; 128: 693-705.].
Understanding the role of histone PTMs is complicated by the fact that the same chemical modification located at different positions within a single protein can have different effects on gene expression. For example, within the histone 3 tail, methylation of lysine 4 (H3K4), H3K36, or H3K79 results in activation of transcription, whereas methylation of H3K9 or H3K27 results in transcriptional repression. This situation is further complicated by the fact that both arginine and lysine can be variably methylated, causing the overall transcriptional outcome to be dictated by both the position of the residue and the degree of methylation [7Martin C, Zhang Y. The diverse functions of histone lysine methylation Nat Rev Mol Cell Biol 2005; 6: 838-49.]. Histone methyltransferases can add up to three methyl groups to a single lysine side chain, while arginine can exist as either monomethyl or dimethyl arginine, the latter in either a symmetric (sRme2) or asymmetric (aRme2) fashion (Fig. 1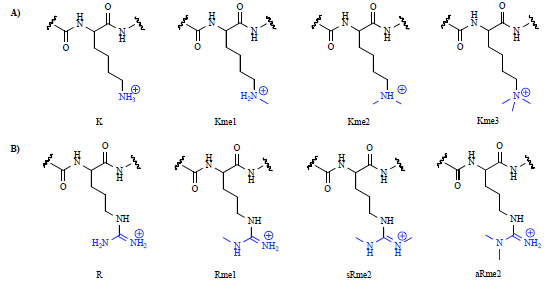 ).
).
 |
Fig. (1) Different methylation states of lysine (A) and arginine (B). |
Although the mechanisms by which cells decipher a PTM-mediated histone code are far from completely understood, it is becoming clear that histone PTMs are read by effector proteins which facilitate downstream events via the recruitment and/or stabilization of chromatin-templated machinery [8Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains Nat Rev Mol Cell Biol 2006; 7: 473-83., 9Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark Mol Cell 2007; 25: 15-30.]. Exploration of these epigenetic readers therefore plays a key role in furthering our understanding of how histone PTMs regulate complex biological functions [10Bottomley MJ. Structures of protein domains that create or recognize histone modifications EMBO Rep 2004; 5: 464-9., 11Daniel JA, Pray-Grant MG, Grant PA. Effector proteins for methylated histones: an expanding family Cell Cycle 2005; 4: 919-26.]. The identification of binding modules for methylated lysines has been largely successful, whereas protein receptors for methylated arginines in histone proteins have received less attention to date [12Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers Nat Struct Mol Biol 2007; 14: 1025-40.]. While methyl-lysine and -arginine readers are most commonly associated with the recognition of histone modifications, they are also known to interact with methylation marks on non-histone proteins, as will be discussed in further detail. The identification of novel reader proteins remains a challenge, as does the broader goal of understanding the relationship between PTM binding proteins and human disease.
Current estimates of the number of methyl-lysine binding proteins in the human proteome exceeds 170 [13Simple Modular Architecture Research Tool (SMART) Database In: 2010.] and this number continues to grow with ongoing research. Despite various structural and functional differences, methyl-lysine and -arginine readers share many common features which facilitate their recognition of these PTMs. All methylated forms of lysine are cationic at physiological pH, while trimethyllysine contains a fixed positive charge irrespective of its environment. As the size, hydrophobicity, distribution of positive charge, and ability to serve as a hydrogen bond donor differs between methylation states, each PTM interacts with a protein reader that can adapt to these specific inherent physical properties. A subtle change in methylation state can impact the resulting protein-protein interaction with profound consequences for gene regulation and expression. A recent publication analyses the effect of the methylation state on one of the effector proteins (L3MBTL1) by means of molecular dynamics and free energy perturbation techniques combined with biophysical binding data in the context of a small-molecule model system [14Gao C, Herold JM, Kireev D, Wigle T, Norris JL, Frye S. Biophysical Probes Reveal a "Compromise" Nature of the Methyl-lysine Binding Pocket in L3MBTL1 J Am Chem Soc 2011; 133: 5357-62.]. Gaining a greater understanding of the atomic-level mechanisms by which methyl-lysine recognition occurs will be helpful in understanding far more complex phenomena including how the effector proteins control many biological processes.
The conserved recognition of methyl-lysine marks is largely mediated by the interaction between the methylammonium group and aromatic residues in the protein receptor, which form an aromatic “cage” around the PTM. Such aromatic cages tend to be relatively specific for a certain methylation state, discriminating between PTMs based on differences in size and shape. The binding interaction between the methylammonium and the aromatic cage is largely the result of cation-π interactions, while hydrophobic desolvation effects also have a substantial role. The cation-π interaction is generally thought of as a charge-quadrupole interaction between a positively charged species and an aromatic ring, primarily electrostatic in nature [15Ma JC, Dougherty DA. The Cation-p Interaction Chem Rev 1997; 97: 1303-24., 16Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology Proc Natl Acad Sci USA 1999; 96: 9459-64.]. The importance of cation-π interactions in the context of proteins has been described previously by Burley and Petsko in 1986 [17Burley SK, Petsko GA. Amino-aromatic interactions in proteins FEBS Lett 1986; 203: 139-43.], and this recognition motif has been seen to be highly conserved in many protein-protein interactions. In the recognition of the lower methylation states, hydrogen bonding and steric exclusion also become increasingly important. Depending on the methylation state, nearby acidic residues in the protein are also known to form salt bridges with the methylated lysine residue, offering an additional stabilizing effect [18Adams-Cioaba MA, Min J. Structure and function of histone methylation binding proteins Biochem Cell Biol 2009; 87: 93-105.]. Based on current understanding, the lower methylation states of lysine (Kme1&2) can be found to bind via a cavity-insertion recognition mode whereby the methylammonium group is deeply buried within the protein while neighboring residues in the histone peptide are making few interactions, causing little sequence selectivity to be observed in vitro. In contrast, trimethyl-lysine (Kme3) is predominantly recognized via surface-groove recognition whereby the peptide lies along the protein surface enabling surrounding residues and the peptide backbone to form additional interactions with the effector protein, leading to a more sequence selective binding event [12Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers Nat Struct Mol Biol 2007; 14: 1025-40.]. Understanding of the different modes of recognition has been and will continue to be significantly advanced with the availability of crystal structures of these domains.
While much attention in this field has been focused on the readers of methylated lysines, arginine methylation has similarly been identified as a key player in the regulation of cellular processes [19McBride AE, Silver PA. State of the arg: protein methylation at arginine comes of age Cell 2001; 106: 5-8.-21Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function Mol Cell 2005; 18: 263-72.]. Although our present knowledge of methyl-arginine effector proteins is limited, there is evidence that methyl-arginine serves as a mediator of protein- protein interactions [22Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code Nat Struct Mol Biol 2004; 11: 1037-43.]. Similarly to lysine, there is precedence for the stacking of arginine with aromatic residues via cation-π interactions [23Burley SK, Petsko GA. Amino-aromatic interactions in proteins FEBS Lett 1986; 203: 139-43.-25Mitchell JB, Nandi CL, McDonald IK, Thornton JM, Price SL. Amino/aromatic interactions in proteins: is the evidence stacked against hydrogen bonding? J Mol Biol 1994; 239: 315-.]. Arginine’s ability to bind to aromatic residues is due to its ability to interact via a combination of cation-π and π-π stacking interactions, and methylation of arginine is thus expected to magnify this interaction.
Despite a conserved cation-π recognition motif across most methyl-lysine and -arginine binding proteins, the protein readers also belong to distinct families based on other specific characteristics. Consideration of the differences between reader families will aid in the development of selective probes and drugs targeting specific proteins. The recognition of methyl-lysine and -arginine residues generally occurs via interaction of a specific sub-domain within the protein. The proteins which are known to contain methyl-lysine binding domains are the plant homeodomain (most commonly referred to as PHD fingers), the Royal Family proteins consisting of Tudor, Agenet, Chromo, PWWP (Proline-Tryptophan-Tryptophan-Proline domains) and MBT (Malignant Brain Tumor) domains, and finally the WD40 repeat protein [26Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP. The Tudor domain 'Royal Family': Tudor, plant Agenet, Chromo, PWWP and MBT domains Trends Biochem Sci 2003; 28: 69-74.]. A recent review of the structural biology of the various PTM reader proteins gives an excellent overview of the structural features and subdomains of these proteins [27Yap KL, Zhou MM. Keeping it in the family: diverse histone recognition by conserved structural folds Crit Rev Biochem Mol Biol 2010; 45(6): 488-505.].
Current drug discovery efforts are largely directed toward the enzymatic proteins which install and remove lysine methylation marks (PTM writers and erasers, respectively) [28Copeland RA, Olhava EJ, Scott MP. Targeting epigenetic enzymes for drug discovery Curr Opin Chem Biol 2010; 14: 505-10.]. In constrast, binding proteins for PTM histone marks have only just emerged as valid targets of probe research and drug discovery. For example, Filippakopoulos and co-workers have recently shown that bromodomain, the readers of acetyl-lysine, can be targeted by small molecule probes [29Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains Nature 2010; 468(7327): 1067-73., 30Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic Nature 2010; 468: 1119-23.]. The reason methyl-lysine readers have received less attention to date is two-fold. First, the readers of methyl-lysine marks are usually characterized by a low affinity for their respective peptide substrates, making any high-throughput assay difficult. Second, their lack of enzymatic activity has hindered their validation as critical drug targets. Small molecule antagonists of reader domains offer the potential of different in vivo efficacy and toxicity as compared to inhibitors of the writer and eraser enzymes [31Taverna SD, Cole PA. Drug discovery: Reader's block Nature 2010; 468: 1050-.] and also can contribute greatly to a growing understanding of how small molecules can target protein-protein interactions. This review will focus primarily on the families of methyl-lysine readers and their potential in the field of drug discovery, while the homologous readers of methyl-arginine will be discussed within the related family of lysine reader.
MBT FAMILY
The malignant brain tumor (MBT) domain is known to recognize the lower methylation states of lysine (Kme1 and Kme2) on both histones and other regulatory proteins. The MBT family is comprised of 9 human members, each of which contains MBT subdomains occurring in tandem repeats of 2-4 units (Fig. 2 ). The MBT domains are often flanked by other subdomains, which in some cases are suggested to assist in dimerization (SPM domain) or support binding to DNA (Zn-finger domain).
). The MBT domains are often flanked by other subdomains, which in some cases are suggested to assist in dimerization (SPM domain) or support binding to DNA (Zn-finger domain).

The MBT domain itself consists of ca. 100 amino acids, and highly conserved homologs can be found in humans, Drosophila, and C. elegans. A recent review of the MBT family highlights its structural features and the cellular functions of each of its members [32Bonasio R, Lecona E, Reinberg D. MBT domain proteins in development and disease Semin Cell Dev Biol 2010; 21: 221-30.]. The interaction between the MBT readers and methylated lysine residues is best described as a cavity-insertion mode [12Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers Nat Struct Mol Biol 2007; 14: 1025-40.] of recognition and the available co-crystal structures for five different domains indicate few interactions beyond the methylated lysine residue. A consequence of this localized interaction is that MBT domains are almost entirely non-sequence selective as long as the methylated peptide shows a high isoelectric point [33Kalakonda N, Fischle W, Boccuni P, et al. Histone H4 lysine 20 monomethylation promotes transcriptional repression by L3MBTL1 Oncogene 2008; 27: 4293-304.]. Most of the MBT proteins can be evolutionary linked to one of three Drosophila orthologs: dL3MBTL, dSCM (Sex Comb on Midleg, Fig. (2B) shows one human ortholog: SCML2), or the four MBT repeat containing dSFMBT (SCM-related gene containing Four MBT domains) [34Klymenko T, Papp B, Fischle W, et al. A Polycomb group protein complex with sequence-specific DNA-binding and selective methyl-lysine-binding activities Genes Dev 2006; 20: 1110-22.].
MBT domains are essential for transcriptional regulation and share an overall conserved binding mechanism. For example, L3MBTL1 has a slight preference for monomethyl-lysine over the dimethyl analog. Its aromatic binding pocket is made up of a tyrosine, phenylalanine, and tryptophan residue with an essential hydrogen bond between the lysine ammonium group and an aspartic acid (D355, Fig. 3A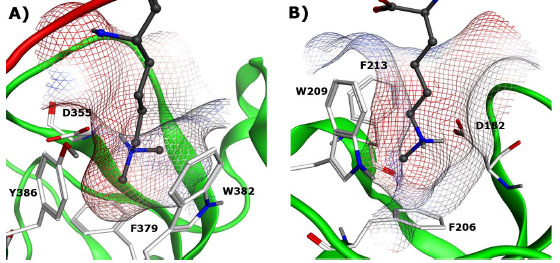 ). In contrast, SCML2 and SCMH1 are the only MBT domains in which the MBT repeat occurs in tandem and exhibits a stronger preference for monomethyl-lysine [35Montini E, Buchner G, Spalluto C, et al. Identification of SCML2, a second human gene homologous to the Drosophila sex comb on midleg (Scm): A new gene cluster on Xp22 Genomics 1999; 58: 65-72.-37Santiveri CM, Lechtenberg BC, Allen MD, Sathyamurthy A, Jaulent AM, Freund SM, et al. The malignant brain tumor repeats of human SCML2 bind to peptides containing monomethylated lysine J Mol Biol 2008; 382: 1107-2.]. In these two reader proteins the binding cavity contains a phenylalanine, replacing the tyrosine in L3MBTL1 (Fig. 3B).
). In contrast, SCML2 and SCMH1 are the only MBT domains in which the MBT repeat occurs in tandem and exhibits a stronger preference for monomethyl-lysine [35Montini E, Buchner G, Spalluto C, et al. Identification of SCML2, a second human gene homologous to the Drosophila sex comb on midleg (Scm): A new gene cluster on Xp22 Genomics 1999; 58: 65-72.-37Santiveri CM, Lechtenberg BC, Allen MD, Sathyamurthy A, Jaulent AM, Freund SM, et al. The malignant brain tumor repeats of human SCML2 bind to peptides containing monomethylated lysine J Mol Biol 2008; 382: 1107-2.]. In these two reader proteins the binding cavity contains a phenylalanine, replacing the tyrosine in L3MBTL1 (Fig. 3B).

Many members of the MBT family and their related biological functions have been studied in detail, however this review will focus on L3MBTL1 and L3MBTL3. The structural biology of L3MBTL1 has been well covered in recent literature [38Min J, Allali-Hassani A, Nady N, et al. L3MBTL1 recognition of mono- and dimethylated histones Nat Struct Mol Biol 2007; 14: 1229-30.], providing a valuable starting point for ligand-based drug design. Furthermore, L3MBTL1 has been shown to act as a chromatin lock [39Trojer P, Li G, Sims RJ, et al. L3MBTL1, a histone-methylation-dependent chromatin lock Cell 2007; 129: 915-28.] and to be important for transcriptional repression [33Kalakonda N, Fischle W, Boccuni P, et al. Histone H4 lysine 20 monomethylation promotes transcriptional repression by L3MBTL1 Oncogene 2008; 27: 4293-304.]. This methyl-lysine reader represses the expression of E2F regulated genes, such as the oncogenic and growth related c-myc gene. As various members of the MBT family are not sequence selective, L3MBTL1 has also been shown to bind the tumor suppressor, p53, through recognition of the methylated lysine residue 382 [40West LE, Roy S, Lachmi-Weiner K, et al. The MBT repeats of L3MBTL1 link set8 mediated p53 methylation at lysine 382 to target gene repression J Biol Chem 2010; 285: 37725-2.]. It has also been suggested that L3MBTL1 might bind to the retinoblastoma (Rb) protein in an analogous fashion, and consequently facilitates repression of c-myc. In addition, in a recent report the Nimer lab has shown that L3MBTL1 positivily influences genomic stability [41Gurvich N, Perna F, Farina A, et al. L3MBTL1 polycomb protein, a candidate tumor suppressor in del(20q12) myeloid disorders, is essential for genome stability Proc Natl Acad Sci USA 2010; 107: 22552-7.], while Perna and co-workers have demonstrated the effect of L3MBTL1 on erythroid differentiation [42Perna F, Gurvich N, Hoya-Arias R, et al. Depletion of L3MBTL1 promotes the erythroid differentiation of human hematopoietic progenitor cells: possible role in 20q- polycythemia vera Blood 2010; 116: 2812-1.].
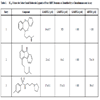
Small molecule ligand discovery for methyl-lysine binding domains is relatively unexplored territory. The first reported virtual screening (VS) study on MBT proteins was carried out by Kireev and co-workers [43Kireev D, Wigle TJ, Norris-Drouin J, Herold JM, Janzen WP, Frye SV. Identification of Non-Peptide Malignant Brain Tumor (MBT) Repeat Antagonists by Virtual Screening of Commercially Available Compounds J Med Chem 2010; 53: 7625-31.]. The iResearch Library (ChemNavigator) which contained more than 50 million procurable compounds was virtually screened for MBT ligands. In this study, two complementary VS approaches were utilized: a substructure search for compounds containing Kme1 and Kme2-‘like’ side chains and a pharmacophore screen followed by docking to search for more structurally remote compounds mimicking the histone peptide interaction. A total of 51 compounds were subsequently purchased and tested against a panel of four MBT-containing proteins (L3MBTL1, L3MBTL3, L3MBTL4 and MBTD1) using an in vitro chemiluminescent assay (AlphaScreen) [44Wigle TJ, Herold JM, Senisterra GA, et al. Screening for inhibitors of low-affinity epigenetic peptide-protein interactions: an AlphaScreen-based assay for antagonists of methyl-lysine binding proteins J Biomol Screen 2010; 15: 62-71.]. Nineteen compounds showed specific dose-dependent protein binding activity and provided inital structure−activity information for lead generation (A selection of these small molecules is shown in Table 1).
More importantly, the first co-crystal structure of L3MBTL1 with a small molecule ligand has recently been published, (Fig. 4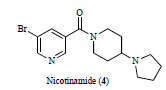 , PDB: 3P8H) along with a detailed study of the binding event using medicinal chemistry, mutagenesis and multiple orthogonal assay formats [45Herold JM, Wigle TJ, Norris JL, et al. Small-molecule ligands of methyl-lysine binding proteins J Med Chem 2011; 54: 2504-11.]. The nicotinamide ligand is shown to bind in the second domain of L3MBTL1 analogously to the native peptides. The significantly larger and more rigid amine anchor makes a critical interaction with the acidic residue D355 and fills out the binding pocket almost entirely (See Fig. 3A). These studies represent the first steps towards high-quality chemical probes for methyl-lysine binding domains though further improvements in potency, greater selectivity profiling and cell-based evidence of activity and mechanism are yet to be addressed.
, PDB: 3P8H) along with a detailed study of the binding event using medicinal chemistry, mutagenesis and multiple orthogonal assay formats [45Herold JM, Wigle TJ, Norris JL, et al. Small-molecule ligands of methyl-lysine binding proteins J Med Chem 2011; 54: 2504-11.]. The nicotinamide ligand is shown to bind in the second domain of L3MBTL1 analogously to the native peptides. The significantly larger and more rigid amine anchor makes a critical interaction with the acidic residue D355 and fills out the binding pocket almost entirely (See Fig. 3A). These studies represent the first steps towards high-quality chemical probes for methyl-lysine binding domains though further improvements in potency, greater selectivity profiling and cell-based evidence of activity and mechanism are yet to be addressed.
 |
Fig. (4) Nicotinamide ligand (4) shown in the co-crystal structure with L3MBTL1. PDB: 3P8H. |
The involvement of MBT domains in differentiation has been seen in similar fashion for L3MBTL3, a close homolog of L3MBTL1. Northcott and co-workers reported the potential involvement of L3MBTL3 in the occurrence of malignant pediatric brain tumors [46Northcott PA, Nakahara Y, Wu X, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma Nat Genet 2009; 41: 465-72.]. High resolution single nucleotide polymorphism (SNP) genotyping identified the amplification and deletion of tumor suppressor genes, including homo- and heterozygous deletions of modules of histone lysine methylation including the readers L3MBTL3, L3MBTL2, and SCML2 and the writers GLP (also known as EHMT1) and SMYD4.
Based on recent literature highlighting the significance of L3MBTL1 and L3MBTL3 in oncogenesis, being able to target these proteins directly would serve as an ideal tool for cancer biology, especially as it is unknown if these deregulated readers are actively promoting oncogenesis or mere bystanders [47Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers Nat Rev Cancer 2010; 10: 457-69.]. Small molecule antagonists of MBT domain interactions could be instrumental to determine, for instance, the role of L3MBTL3 in the development of medulloblastomas, which are still the leading cause of cancer-related deaths in children.
PHD FINGERS
The plant homeodomain, also referred to as PHD finger, is a structural motif consisting of approximately 50 to 80 amino acids. The PHD finger family is one of the largest and most diverse of the methyl-lysine readers, and within this family two general classes have been identified based on their preferred binding partner: the first group binds di- and trimethylated lysine residues whereas the second group interacts with unmethylated lysine residues. PHD fingers which bind unmodified lysine residues apparently organize their respective binding pockets only in the presence of their binding partner, while the binders of di- and trimethyl-lysine are known to have a preorganized binding pocket, similar to the previously mentioned MBT domains [48Wang Z, Song J, Milne TA, et al. Pro isomerization in MLL1 PHD3-bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression Cell 2010; 141: 1183-94.]. Some of the PHD fingers are good examples of surface-groove recognition readers which was discussed above. The extended interaction of the methylated lysine and neighboring residues leads to selectivity for a specific PTM in those cases. For drug design these additional interactions could also prove crucial in addressing the questions of selectivity and potency. It is also worth noting that for any probe design or drug discovery efforts targeting the readers of trimethyl-lysine such as the PHD fingers or other domains discussed below, an uncharged substitute for the trimethylammonium group is required to achieve cell-permeability.
Of the 81 known members of the family, a significant number have structural data available in the form of crystal structures (13) and NMR structures (14), both of which will be of great value in future efforts focused on the design of chemical probes. The ability of PHD fingers to bind both unmethylated and methylated lysine residues demonstrates the diversity among this family, and it is worthwhile to highlight distinct members of the PHD family to illustrate their structural biology (Fig. 5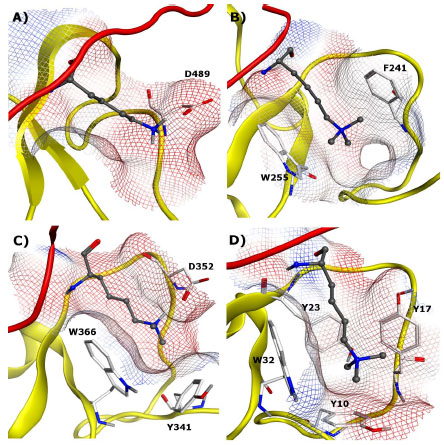 ) and function.
) and function.

PHF21A (BHC80) specifically interacts with unmethylated H3K4 (H3K4me0) (Fig. 5A) [49Lan F, Collins RE, De Cegli R, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression Nature 2007; 448: 718-22.]. Unlike most other reader proteins where the binding pockets feature an aromatic cage, there are no aromatic residues inside the binding pocket of PHF21A (Fig. 5A). The crystal structure revealed that the specificity for H3K4me0 is achieved through the hydrogen bond formed between the lysine ε-amine and D489 of the binding pocket, and further stabilized by an additional hydrogen bond between the amine and the E488 backbone. The structure also suggests that steric exclusion of the methyl group prevents methylated H3K4 from binding to this pocket. Knockdown of PHF21A by RNA inhibition results in the de-repression of LSD1 target genes, and this repression is restored by the reintroduction of wild-type PHF21A but not by the D489A mutant, which does not bind H3K4. These findings highlight the importance of PHF21A in gene repression, and more interestingly, suggests unmethylated lysine residues are subject to specific reader interactions in the absence of PTM.
The importance of PHD fingers in human disease was recently outlined in a review by Baker and co-workers [50Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks Mutat Res 2008; 647: 3-12.]. In many cases, PHD fingers are linked to disease as a consequence of mutants or translocations of the native proteins. In such cases, drug discovery efforts focused towards such mutant proteins rather than the wildtype proteins would likely be most effective. However, a few examples are known where PHD fingers are associated with human disease and could be targeted directly. For example, Wang and co-workers have reported that a fusion protein containing a PHD finger (PHF23) and Nucleoporin (NUP98) showed all the characteristics of a potent oncoprotein whose function was dependent on binding to H3K4me3 [51Wang GG, Song J, Wang Z, et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger Nature 2009; 459: 847-51.]. The phenotypic consequences of this mutation included arrested haematopoietic differentiation and acute myeloid leukemia. PHF23 has very high sequence homology with PHF13 (Fig. 5B), for which a crystal structure has been solved.
Besides the potential to combat cancer in innovative ways, the readers of methyl-lysine residues have also been shown to play a role in differentiation and stem cell self-renewal. Targeting these proteins may therefore open new avenues for the generation and maintenance of stem cells and advance the field of regenerative medicine. Walker and co-workers have recently reported that the interaction between polycomb-like protein 2 (PCL2) and the polycomb repressive complex 2 (PRC2) is important for de-differentiation and self-renewal [52Walker E, Chang WY, Hunkapiller J, et al. Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation Cell Stem Cell 2010; 6: 153-66.]. Interestingly enough, PCL2 contains a Tudor domain (discussed below) and two PHD fingers which are necessary to bind H3K27me3 and in turn direct PRC2 function to specific targets.
TUDOR DOMAINS
Tudor domains belong to the Royal superfamily of methyl-lysine effector proteins, and consist of structurally diverse proteins which display a range of recognition motifs, interacting with both higher and lower lysine methylation states. There are currently close to 40 known Tudor containing proteins, with 12 available crystal structures [53Structural Genomics Consortium (SGC) Available from: www.thesgc.org 2011.]. Importantly, it has been reported that the Tudor family of proteins are closely linked to gametogenesis, in both Drosophila and mice, while also playing roles in various piRNA pathways [54Siomi MC, Mannen T, Siomi H. How does the royal family of Tudor rule the PIWI-interacting RNA pathway? Genes Dev 2010; 24: 636-46.]. As there is not a complete understanding of the role of Tudor domains as methyl-lysine and -arginine readers, the development of small molecule modulators of these protein-protein interactions would serve as a valuable tool in developing a more thorough understanding of the role of Tudor domains in both disease and development.
Tudor domains are characterized by a bent antiparallel β-barrel, with conserved residues stabilizing the structure through formation of a hydrophobic core and an overall negatively charged surface [55Selenko P, Sprangers R, Stier G, Buhler D, Fischer U, Sattler M. SMN tudor domain structure and its interaction with the Sm proteins Nat Struct Biol 2001; 8: 27-31.]. Tudor proteins which recognize methyl-lysine are generally classified as tandem or double tudor domains, while a single Tudor domain methyl-lysine reader has yet to be identified. JMJD2A is a well known double Tudor domain which functions as both a demethylase and methyl-lysine binding protein (Fig. 6A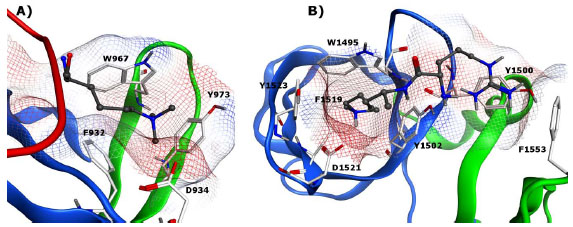 ). The reader contains an interdigitated folding of two individual Tudor domains, linked by two shared β-strands. It is this interleaved, bilobal topology of the two domains which is required to form an appropriate binding pocket to interact with methylated histone H3K4 and H4K20 [56Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A Science 2006; 312: 748-51.]. Two aromatic residues from the second tudor sequence and one from the first tudor sequence generate an aromatic cage, which in conjunction with a nearby aspartate residue form a binding pocket for trimethyllysine. In contrast to the double Tudor domains of JMJD2A, mammalian p53-binding protein (53BP1) has been shown to recognize the lower lysine methylation states of both p53 and H4K20 [57Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage Cell 2004; 119: 603-14., 58Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint Science 2002; 298: 1435-8.]. The ability of 53BP1 to bind both H4K20me2 and p53K382me2 is important as it facilitates p53 recruitment to DNA damage sites, where the H4K20me2 modification is prevalent, thereby promoting DNA double-strand break repair [59Roy S, Musselman CA, Kachirskaia I, et al. Structural insight into p53 recognition by the 53BP1 tandem Tudor domain J Mol Biol 2010; 398: 489-96.]. The structure of 53BP1 differs appreciably from that of JMJD2A despite significant sequence homology; 53BP1 consists of two independently folded tudor domains, of which the first is primarily involved in methyl lysine recognition assisted by the second (Fig. 6B). The specificity of 53BP1 for Kme1 and Kme2 is the consequence of both an intermolecular hydrogen bond between the mono- or dimethylammonium group and an aspartic acid residue located in the central binding pocket and the fact that it is apparently not large enough to accommodate a trimethyl-lysine mark [60Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair Cell 2006; 127: 1361-73.]. In addition, 53BP1 is shown to recognize a neighboring basic residue such as an arginine (H4R19) (Fig. 6B) or a lysine (p53K381) in a second aromatic pocket.
). The reader contains an interdigitated folding of two individual Tudor domains, linked by two shared β-strands. It is this interleaved, bilobal topology of the two domains which is required to form an appropriate binding pocket to interact with methylated histone H3K4 and H4K20 [56Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A Science 2006; 312: 748-51.]. Two aromatic residues from the second tudor sequence and one from the first tudor sequence generate an aromatic cage, which in conjunction with a nearby aspartate residue form a binding pocket for trimethyllysine. In contrast to the double Tudor domains of JMJD2A, mammalian p53-binding protein (53BP1) has been shown to recognize the lower lysine methylation states of both p53 and H4K20 [57Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage Cell 2004; 119: 603-14., 58Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint Science 2002; 298: 1435-8.]. The ability of 53BP1 to bind both H4K20me2 and p53K382me2 is important as it facilitates p53 recruitment to DNA damage sites, where the H4K20me2 modification is prevalent, thereby promoting DNA double-strand break repair [59Roy S, Musselman CA, Kachirskaia I, et al. Structural insight into p53 recognition by the 53BP1 tandem Tudor domain J Mol Biol 2010; 398: 489-96.]. The structure of 53BP1 differs appreciably from that of JMJD2A despite significant sequence homology; 53BP1 consists of two independently folded tudor domains, of which the first is primarily involved in methyl lysine recognition assisted by the second (Fig. 6B). The specificity of 53BP1 for Kme1 and Kme2 is the consequence of both an intermolecular hydrogen bond between the mono- or dimethylammonium group and an aspartic acid residue located in the central binding pocket and the fact that it is apparently not large enough to accommodate a trimethyl-lysine mark [60Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair Cell 2006; 127: 1361-73.]. In addition, 53BP1 is shown to recognize a neighboring basic residue such as an arginine (H4R19) (Fig. 6B) or a lysine (p53K381) in a second aromatic pocket.

Although the characterization of Tudor domains as methyl-arginine readers is less well documented, there is substantial evidence that the SMN (Survival of Motor Neuron) Tudor domain, which is linked to spinal muscular atrophy, recognizes the arginine-glycine rich C-terminal tails of spliceosomal Sm proteins and that this binding event is mediated by symmetrical dimethylation of arginine side chains [61Sprangers R, Groves MR, Sinning I, Sattler M. High-resolution X-ray and NMR structures of the SMN Tudor domain: conformational variation in the binding site for symmetrically dimethylated arginine residues J Mol Biol 2003; 327: 507-20., 62Cote J, Richard S. Tudor domains bind symmetrical dimethylated arginines J Biol Chem 2005; 280: 28476-83.]. It is proposed that this interaction is facilitated by the positioning of a symmetrical dimethylarginine side chain near a cluster of conserved aromatic residues, forming a typical cage-like mode of recognition. In another case, it has been demonstrated that members of the Tudor family associate with PIWI (P-element-induced wimpy testis) specifically through sRme2 [54Siomi MC, Mannen T, Siomi H. How does the royal family of Tudor rule the PIWI-interacting RNA pathway? Genes Dev 2010; 24: 636-46., 63Liu K, Chen C, Guo Y, et al. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain Proc Natl Acad Sci USA 2010; 107: 18398-403.]. More recently, it was shown that Tudor protein human SND1 (staphylococcal nuclease domain-containing 1) binds PIWIL1 in an arginine methylation dependent manner, suggesting a previously undescribed function for SND1 in regulating piRNA pathways [63Liu K, Chen C, Guo Y, et al. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain Proc Natl Acad Sci USA 2010; 107: 18398-403.]. Crystal structures revealed that the intact SND1 extended Tudor domain forms a wide and negatively charged binding groove which can appropriately accommodate sRme2 peptides from PIWIL1 in different orientations [63Liu K, Chen C, Guo Y, et al. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain Proc Natl Acad Sci USA 2010; 107: 18398-403.].
CHROMODOMAINS
The chromodomain is a highly conserved family of methyl-lysine reader proteins found in both plants and animals, consisting of 40-50 amino acids and spanning 34 known members [53Structural Genomics Consortium (SGC) Available from: www.thesgc.org 2011.]. One of the earliest and best-known protein-protein interactions induced by methyl-lysine recognition is the binding of H3K9me3 to the HP1 (heterochromatin-associated protein 1) chromodomain, which in turn results in gene silencing. HP1 and other members of the chromodomain family are generally known to consist of an N-terminal three-stranded anti-parallel β-sheet which folds against a C-terminal α-helix [64Ball LJ, Murzina NV, Broadhurst RW, et al. Structure of the chromatin binding (chromo) domain from mouse modifier protein 1 EMBO J 1997; 16: 2473-81.]. For HP1, the binding affinities have been reported to be in the low micromolar range for both H3K9me3 and H3K9me2 [65Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail Science 2002; 295: 2080-3., 66Hughes RM, Wiggins KR, Khorasanizadeh S, Waters ML. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect Proc Natl Acad Sci USA 2007; 104: 11184-8.]. The aromatic binding cage is made up of three residues which form a conserved aromatic pocket into which the methylammonium group inserts itself (see Fig. 7A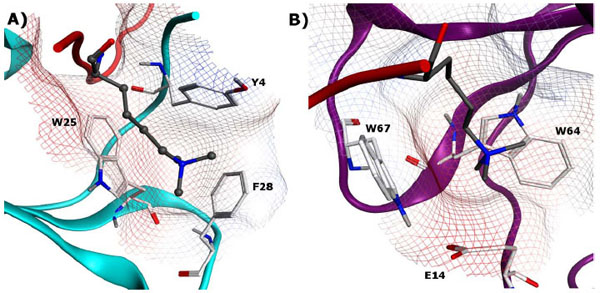 for human homolog of HP1). Mutation of any of these aromatic residues drastically reduces affinity for the methylated histone tail. Furthermore, residues 5-10 of the histone tail (QTARK9S) interact with the chromodomain by an induced-fit sandwiching between terminal β-strands, completing a five-stranded antiparallel β-sheet [65Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail Science 2002; 295: 2080-3.]. Mutation studies of residues in both the peptide and protein have confirmed the contribution of intermolecular contacts along the extended surface groove to both binding affinity and selectivity [67Nielsen PR, Nietlispach D, Mott HR, et al. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9 Nature 2002; 416: 103-7.].
for human homolog of HP1). Mutation of any of these aromatic residues drastically reduces affinity for the methylated histone tail. Furthermore, residues 5-10 of the histone tail (QTARK9S) interact with the chromodomain by an induced-fit sandwiching between terminal β-strands, completing a five-stranded antiparallel β-sheet [65Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail Science 2002; 295: 2080-3.]. Mutation studies of residues in both the peptide and protein have confirmed the contribution of intermolecular contacts along the extended surface groove to both binding affinity and selectivity [67Nielsen PR, Nietlispach D, Mott HR, et al. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9 Nature 2002; 416: 103-7.].

In comparison to HP1, methyl-lysine recognition by chromobox (CBX) proteins (for example, Polycomb) involve fewer contacts with the residues surrounding the methyl-lysine [68Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains Genes Dev 2003; 17: 1870-81.]. While this is often associated with a decrease in sequence selectivity, it can be envisioned that such non-sequence selective recognition domains are more amenable to inhibition by small molecules due to their limited binding sites. Furthermore, tandem chromodomains have been reported such as CHD (chromo helicase DNA-binding) proteins in which the two chromodomains are bridged by a two-helix linker to form a continuous surface. The human CHD1 double chromodomain, for example, interacts with H3K4 methylation sites, a hallmark of active chromatin. The two CHD1 chromodomains are seen to cooperatively interact with one methylated H3 tail at the chromodomain junction, using only two aromatic residues for methyl-lysine recognition in contrast to the 3-residue aromatic cage of HP1 (Fig. 7B) [69Flanagan JF, Mi LZ, Chruszcz M, et al. Double chromodomains cooperate to recognize the methylated histone H3 tail Nature 2005; 438: 1181-5.].
While ongoing research is helping to elucidate the binding sites and interacting proteins of various chromodomain-containing proteins, it is also becoming increasingly clear that mutations in such domains are closely linked to a variety of disorders. In 2004, Vissers and co-workers identified that heterozygous mutations in Chromodomain Helicase DNA-binding protein 7 (CHD7) cause the CHARGE syndrome [70Pagon RA, Graham JM, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association J Pediatr 1981; 99: 223-7., 71Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome Nat Genet 2004; 36: 955-7.] (Coloboma of the eye, Heart defects, Atresia of the choanae, severe Retardation of growth and development, Genital abnormalities, and Ear abnormalities), a disease with an estimated incidence of approximately 1 in 10,000 newborns. In a recent study combining Chromatin Immunoprecipiation with microarray technology (ChIP-chip), the role of CHD7 in gene expression was identified. CHD7 was shown to bind to mono- and dimethylated histone H3K4 in enhancer regions of numerous genes in a highly cell type-dependent manner. It was observed that CHD7 localization sites change concomitantly with H3K4me patterns during cell differentiation, indicating that the H3K4 methylation mark defines lineage-specific association of CHD7 with specific sites on chromatin [72Schnetz MP, Bartels CF, Shastri K, et al. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns Genome Res 2009; 19: 590-601.]. A selective small molecule ligand of CHD7 could therefore serve as a tool to elucidate the biology behind CHARGE syndrome.
PWWP DOMAINS
The proline-tryptophan-tryptophan-proline (PWWP) motif is a structural domain which recognizes various methylation states of lysine residues and can be found in both proteins as well as enzymes involved in chromatin biology. The PWWP domain of Brpf1 was recently characterized by both NMR and crystallization studies, elucidating both the apo form of the methyl-lysine reader as well as the protein in complex with the H3K36me3 peptide (Fig. 8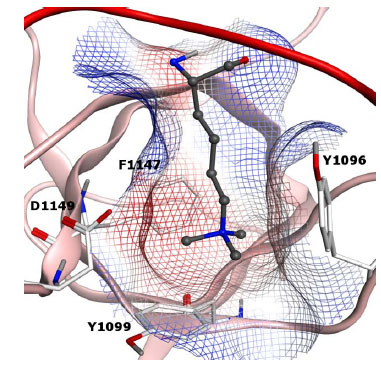 ) [73Vezzoli A, Bonadies N, Allen MD, et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1 Nat Struct Mol Biol 2010; 17: 617-9.].
) [73Vezzoli A, Bonadies N, Allen MD, et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1 Nat Struct Mol Biol 2010; 17: 617-9.].

The PWWP domain is a member of the Royal family and is instrumental for the assembly of a complex involved in acute myeloid and mixed-lineage leukemia (ALL and MLL, respectively) after chromosomal translocations [74Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes Cancer Cell 2008; 14: 36-46.]. As mentioned before with regards to the PHD family, targeting such oncogenic fusion proteins could prove significant for drug discovery (Fig. 8) [29Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains Nature 2010; 468(7327): 1067-73.].
WD40 REPEAT
The WD40 repeat, also known as the beta-transducin repeat, is a protein domain found in numerous readers and enzymes. The first WD40 repeat reported to bind histone modifications is the WDR5 domain found in the MLL/SET1 methyltransferase complex, which binds di- or trimethylation states of H3K4 [75Wysocka J, Swigut T, Milne TA, et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development Cell 2005; 121: 859-72.]. Recently, a second member of the WD40 repeat, EED, was reported to associate with and regulate the activity and specificity of the polycomb repressive complex (PRC2) [76Xu C, Bian C, Yang W, et al. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2) Proc Natl Acad Sci U S A 2010; 107: 19266-71.]. Xu and co-workers demonstrate that the reader protein directly modulates the methyltransferase activity through its binding of various methylated peptides. By using different trimethylated peptides the authors show that H3K27me3 stimulates the transferase activity relative to other trimethylated peptides. Interestingly, EED itself can become methylated by PRC2 when H1K26Me3 is the ligand (Fig. 9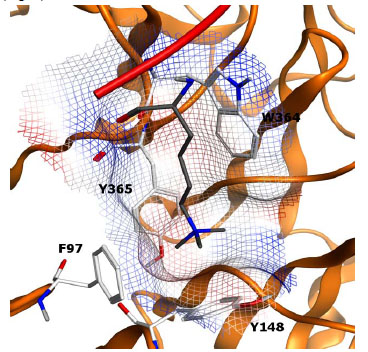 ).
).

DRUG DISCOVERY ENABLED BY CHEMICAL PROBES
The readers of PTMs associated with chromatin regulation currently represent unexplored territory for drug discovery. In order for these protein classes to become part of the druggable genome [77Hopkins AL, Groom CR. The druggable genome Nat Rev Drug Discov 2002; 1: 727-30.] both their tractability and validity need to be experimentally verified. Recent reports of the discovery of potent and selective, cellularly active, small molecule antagonists of acetyl-lysine recognition [29Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains Nature 2010; 468(7327): 1067-73., 30Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic Nature 2010; 468: 1119-23.] targeting the bromodomain BET subfamily, provide significant encouragement that both these issues can be addressed for at least some of the readers of the histone code. Our strategy is to take a protein-family approach [77Hopkins AL, Groom CR. The druggable genome Nat Rev Drug Discov 2002; 1: 727-30.] toward the therapeutically unbiased discovery of high quality chemical probes for readers of methyl-lysine [78Frye SV, Heightman T, Jin J. Targeting Methyl Lysine In: Macor EM, Ed. Annual Reports Medicinal Chemistry. USA: Academic Press 2010; pp. 329-43., 79Frye SV. The art of the chemical probe Nat Chem Biol 2010; 6: 159-61.]. Initial results of ligand discovery via experimental and virtual screening [43Kireev D, Wigle TJ, Norris-Drouin J, Herold JM, Janzen WP, Frye SV. Identification of Non-Peptide Malignant Brain Tumor (MBT) Repeat Antagonists by Virtual Screening of Commercially Available Compounds J Med Chem 2010; 53: 7625-31., 44Wigle TJ, Herold JM, Senisterra GA, et al. Screening for inhibitors of low-affinity epigenetic peptide-protein interactions: an AlphaScreen-based assay for antagonists of methyl-lysine binding proteins J Biomol Screen 2010; 15: 62-71., 80Campagna-Slater V, Schapira M. Finding inspiration in the protein data bank to chemically antagonize readers of the histone code Mol Inf 2010; 29: 322-1.] have confirmed some of the anticipated challenges in terms of potency for these weakly interacting domains, but also show promise for selectivity even within related MBT domain containing proteins (Table 1). By taking a broad and unbiased approach to probe discovery, the chances for finding potency enhancing features in ligands is increased via testing of each ligand hypothesis versus a large number of functionally homologous, but structurally distinct binding sites (as reviewed above). In addition, this approach naturally annotates ligand selectivity and creates structure activity relationships across methyl-lysine reader families. High quality probes for methyl-lysine binders will have utility in exploring the biology of this large and diverse family and will lay the foundation for drug discovery as validated and tractable targets are revealed.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGEMENTS
This work is supported by NIH grant number RC1GM090732. Post-doctoral fellowships for JMH, LAI and CG from the Carolina Partnership are gratefully acknowledged. The authors also acknowledge an enabling collaboration with the Structural Genomics Consortium (SGC).
REFERENCES
| [1] | Bhaumik SR, Smith E, Shilatifard A. Covalent modifications of histones during development and disease pathogenesis Nat Struct Mol Biol 2007; 14: 1008-6. |
| [2] | Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, Part I: Covalent histone modifications Trends Mol Med 2007; 13: 363-72. |
| [3] | Jenuwein T, Allis CD. Translating the histone code Science 2001; 293: 1074-80. |
| [4] | Strahl BD, Allis CD. The language of covalent histone modifications Nature 2000; 403: 41-5. |
| [5] | Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails Genes Dev 2001; 15: 2343-60. |
| [6] | Kouzarides T. Chromatin modifications and their function Cell 2007; 128: 693-705. |
| [7] | Martin C, Zhang Y. The diverse functions of histone lysine methylation Nat Rev Mol Cell Biol 2005; 6: 838-49. |
| [8] | Seet BT, Dikic I, Zhou MM, Pawson T. Reading protein modifications with interaction domains Nat Rev Mol Cell Biol 2006; 7: 473-83. |
| [9] | Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark Mol Cell 2007; 25: 15-30. |
| [10] | Bottomley MJ. Structures of protein domains that create or recognize histone modifications EMBO Rep 2004; 5: 464-9. |
| [11] | Daniel JA, Pray-Grant MG, Grant PA. Effector proteins for methylated histones: an expanding family Cell Cycle 2005; 4: 919-26. |
| [12] | Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers Nat Struct Mol Biol 2007; 14: 1025-40. |
| [13] | Simple Modular Architecture Research Tool (SMART) Database In: 2010. |
| [14] | Gao C, Herold JM, Kireev D, Wigle T, Norris JL, Frye S. Biophysical Probes Reveal a "Compromise" Nature of the Methyl-lysine Binding Pocket in L3MBTL1 J Am Chem Soc 2011; 133: 5357-62. |
| [15] | Ma JC, Dougherty DA. The Cation-p Interaction Chem Rev 1997; 97: 1303-24. |
| [16] | Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology Proc Natl Acad Sci USA 1999; 96: 9459-64. |
| [17] | Burley SK, Petsko GA. Amino-aromatic interactions in proteins FEBS Lett 1986; 203: 139-43. |
| [18] | Adams-Cioaba MA, Min J. Structure and function of histone methylation binding proteins Biochem Cell Biol 2009; 87: 93-105. |
| [19] | McBride AE, Silver PA. State of the arg: protein methylation at arginine comes of age Cell 2001; 106: 5-8. |
| [20] | Lee DY, Teyssier C, Strahl BD, Stallcup MR. Role of protein methylation in regulation of transcription Endocr Rev 2005; 26: 147-70. |
| [21] | Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function Mol Cell 2005; 18: 263-72. |
| [22] | Cosgrove MS, Boeke JD, Wolberger C. Regulated nucleosome mobility and the histone code Nat Struct Mol Biol 2004; 11: 1037-43. |
| [23] | Burley SK, Petsko GA. Amino-aromatic interactions in proteins FEBS Lett 1986; 203: 139-43. |
| [24] | Flocco MM, Mowbray SL. Planar stacking interactions of arginine and aromatic side-chains in proteins J Mol Biol 1994; 235: 709-17. |
| [25] | Mitchell JB, Nandi CL, McDonald IK, Thornton JM, Price SL. Amino/aromatic interactions in proteins: is the evidence stacked against hydrogen bonding? J Mol Biol 1994; 239: 315-. |
| [26] | Maurer-Stroh S, Dickens NJ, Hughes-Davies L, Kouzarides T, Eisenhaber F, Ponting CP. The Tudor domain 'Royal Family': Tudor, plant Agenet, Chromo, PWWP and MBT domains Trends Biochem Sci 2003; 28: 69-74. |
| [27] | Yap KL, Zhou MM. Keeping it in the family: diverse histone recognition by conserved structural folds Crit Rev Biochem Mol Biol 2010; 45(6): 488-505. |
| [28] | Copeland RA, Olhava EJ, Scott MP. Targeting epigenetic enzymes for drug discovery Curr Opin Chem Biol 2010; 14: 505-10. |
| [29] | Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains Nature 2010; 468(7327): 1067-73. |
| [30] | Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic Nature 2010; 468: 1119-23. |
| [31] | Taverna SD, Cole PA. Drug discovery: Reader's block Nature 2010; 468: 1050-. |
| [32] | Bonasio R, Lecona E, Reinberg D. MBT domain proteins in development and disease Semin Cell Dev Biol 2010; 21: 221-30. |
| [33] | Kalakonda N, Fischle W, Boccuni P, et al. Histone H4 lysine 20 monomethylation promotes transcriptional repression by L3MBTL1 Oncogene 2008; 27: 4293-304. |
| [34] | Klymenko T, Papp B, Fischle W, et al. A Polycomb group protein complex with sequence-specific DNA-binding and selective methyl-lysine-binding activities Genes Dev 2006; 20: 1110-22. |
| [35] | Montini E, Buchner G, Spalluto C, et al. Identification of SCML2, a second human gene homologous to the Drosophila sex comb on midleg (Scm): A new gene cluster on Xp22 Genomics 1999; 58: 65-72. |
| [36] | Sathyamurthy A, Allen MD, Murzin AG, Bycroft M. Crystal structure of the malignant brain tumor (MBT) repeats in Sex Comb on Midleg-like 2 (SCML2) J Biol Chem 2003; 278: 46968-73. |
| [37] | Santiveri CM, Lechtenberg BC, Allen MD, Sathyamurthy A, Jaulent AM, Freund SM, et al. The malignant brain tumor repeats of human SCML2 bind to peptides containing monomethylated lysine J Mol Biol 2008; 382: 1107-2. |
| [38] | Min J, Allali-Hassani A, Nady N, et al. L3MBTL1 recognition of mono- and dimethylated histones Nat Struct Mol Biol 2007; 14: 1229-30. |
| [39] | Trojer P, Li G, Sims RJ, et al. L3MBTL1, a histone-methylation-dependent chromatin lock Cell 2007; 129: 915-28. |
| [40] | West LE, Roy S, Lachmi-Weiner K, et al. The MBT repeats of L3MBTL1 link set8 mediated p53 methylation at lysine 382 to target gene repression J Biol Chem 2010; 285: 37725-2. |
| [41] | Gurvich N, Perna F, Farina A, et al. L3MBTL1 polycomb protein, a candidate tumor suppressor in del(20q12) myeloid disorders, is essential for genome stability Proc Natl Acad Sci USA 2010; 107: 22552-7. |
| [42] | Perna F, Gurvich N, Hoya-Arias R, et al. Depletion of L3MBTL1 promotes the erythroid differentiation of human hematopoietic progenitor cells: possible role in 20q- polycythemia vera Blood 2010; 116: 2812-1. |
| [43] | Kireev D, Wigle TJ, Norris-Drouin J, Herold JM, Janzen WP, Frye SV. Identification of Non-Peptide Malignant Brain Tumor (MBT) Repeat Antagonists by Virtual Screening of Commercially Available Compounds J Med Chem 2010; 53: 7625-31. |
| [44] | Wigle TJ, Herold JM, Senisterra GA, et al. Screening for inhibitors of low-affinity epigenetic peptide-protein interactions: an AlphaScreen-based assay for antagonists of methyl-lysine binding proteins J Biomol Screen 2010; 15: 62-71. |
| [45] | Herold JM, Wigle TJ, Norris JL, et al. Small-molecule ligands of methyl-lysine binding proteins J Med Chem 2011; 54: 2504-11. |
| [46] | Northcott PA, Nakahara Y, Wu X, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma Nat Genet 2009; 41: 465-72. |
| [47] | Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers Nat Rev Cancer 2010; 10: 457-69. |
| [48] | Wang Z, Song J, Milne TA, et al. Pro isomerization in MLL1 PHD3-bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression Cell 2010; 141: 1183-94. |
| [49] | Lan F, Collins RE, De Cegli R, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression Nature 2007; 448: 718-22. |
| [50] | Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: disorders arising from misinterpreting epigenetic marks Mutat Res 2008; 647: 3-12. |
| [51] | Wang GG, Song J, Wang Z, et al. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger Nature 2009; 459: 847-51. |
| [52] | Walker E, Chang WY, Hunkapiller J, et al. Polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation Cell Stem Cell 2010; 6: 153-66. |
| [53] | Structural Genomics Consortium (SGC) Available from: www.thesgc.org 2011. |
| [54] | Siomi MC, Mannen T, Siomi H. How does the royal family of Tudor rule the PIWI-interacting RNA pathway? Genes Dev 2010; 24: 636-46. |
| [55] | Selenko P, Sprangers R, Stier G, Buhler D, Fischer U, Sattler M. SMN tudor domain structure and its interaction with the Sm proteins Nat Struct Biol 2001; 8: 27-31. |
| [56] | Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A Science 2006; 312: 748-51. |
| [57] | Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage Cell 2004; 119: 603-14. |
| [58] | Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint Science 2002; 298: 1435-8. |
| [59] | Roy S, Musselman CA, Kachirskaia I, et al. Structural insight into p53 recognition by the 53BP1 tandem Tudor domain J Mol Biol 2010; 398: 489-96. |
| [60] | Botuyan MV, Lee J, Ward IM, et al. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair Cell 2006; 127: 1361-73. |
| [61] | Sprangers R, Groves MR, Sinning I, Sattler M. High-resolution X-ray and NMR structures of the SMN Tudor domain: conformational variation in the binding site for symmetrically dimethylated arginine residues J Mol Biol 2003; 327: 507-20. |
| [62] | Cote J, Richard S. Tudor domains bind symmetrical dimethylated arginines J Biol Chem 2005; 280: 28476-83. |
| [63] | Liu K, Chen C, Guo Y, et al. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain Proc Natl Acad Sci USA 2010; 107: 18398-403. |
| [64] | Ball LJ, Murzina NV, Broadhurst RW, et al. Structure of the chromatin binding (chromo) domain from mouse modifier protein 1 EMBO J 1997; 16: 2473-81. |
| [65] | Jacobs SA, Khorasanizadeh S. Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail Science 2002; 295: 2080-3. |
| [66] | Hughes RM, Wiggins KR, Khorasanizadeh S, Waters ML. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect Proc Natl Acad Sci USA 2007; 104: 11184-8. |
| [67] | Nielsen PR, Nietlispach D, Mott HR, et al. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9 Nature 2002; 416: 103-7. |
| [68] | Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains Genes Dev 2003; 17: 1870-81. |
| [69] | Flanagan JF, Mi LZ, Chruszcz M, et al. Double chromodomains cooperate to recognize the methylated histone H3 tail Nature 2005; 438: 1181-5. |
| [70] | Pagon RA, Graham JM, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association J Pediatr 1981; 99: 223-7. |
| [71] | Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome Nat Genet 2004; 36: 955-7. |
| [72] | Schnetz MP, Bartels CF, Shastri K, et al. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns Genome Res 2009; 19: 590-601. |
| [73] | Vezzoli A, Bonadies N, Allen MD, et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1 Nat Struct Mol Biol 2010; 17: 617-9. |
| [74] | Yokoyama A, Cleary ML. Menin critically links MLL proteins with LEDGF on cancer-associated target genes Cancer Cell 2008; 14: 36-46. |
| [75] | Wysocka J, Swigut T, Milne TA, et al. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development Cell 2005; 121: 859-72. |
| [76] | Xu C, Bian C, Yang W, et al. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2) Proc Natl Acad Sci U S A 2010; 107: 19266-71. |
| [77] | Hopkins AL, Groom CR. The druggable genome Nat Rev Drug Discov 2002; 1: 727-30. |
| [78] | Frye SV, Heightman T, Jin J. Targeting Methyl Lysine In: Macor EM, Ed. Annual Reports Medicinal Chemistry. USA: Academic Press 2010; pp. 329-43. |
| [79] | Frye SV. The art of the chemical probe Nat Chem Biol 2010; 6: 159-61. |
| [80] | Campagna-Slater V, Schapira M. Finding inspiration in the protein data bank to chemically antagonize readers of the histone code Mol Inf 2010; 29: 322-1. |