- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
A Novel Bioluminescent Protease Assay Using Engineered Firefly Luciferase
Susan S Wigdal, Jessica L Anderson, Gediminas J Vidugiris, John Shultz, Keith V Wood, Frank Fan*
Abstract
Proteases play important roles in a variety of disease processes. Understanding their biological functions underpins the efforts of drug discovery. We have developed a bioluminescent protease assay using a circularly permuted form of firefly luciferase, wherein the native enzyme termini were joined by a peptide containing a protease site of interest. Protease cleavage of these mutant luciferases greatly activates the enzyme, typically over 100 fold. The mutant luciferase substrates are easily generated by molecular cloning and cell-free translation reactions and thus the protease substrates do not need to be chemically synthesized or purchased. The assay has broad applicability using a variety of proteases and their cognate sites and can sensitively detect protease activity. In this report we further demonstrate its utility for the evaluation of protease recognition sequence specificity and subsequent establishment of an optimized assay for the identification and characterization of protease inhibitors using high throughput screening.
Article Information
Identifiers and Pagination:
Year: 2008Volume: 2
First Page: 16
Last Page: 28
Publisher Id: CCGTM-2-16
DOI: 10.2174/1875397300802010016
Article History:
Received Date: 20/5/2008Revision Received Date: 20/8/2008
Acceptance Date: 23/8/2008
Electronic publication date: 17/10/2008
Collection year: 2008
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Promega Corporation, 2800 Woods Hollow Road, Madison, WI 53711, USA; Tel: 608-277-2531; Fax: 608-298-4818; E-mail: frank.fan@promega.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 20-5-2008 |
Original Manuscript | A Novel Bioluminescent Protease Assay Using Engineered Firefly Luciferase | |
INTRODUCTION
Proteases are a diverse group of enzymes which hydrolyze peptide bonds. They are essential for many physiological processes including cell proliferation, cell differentiation, tissue remodeling, immune response, complement system, neuronal outgrowth, angiogenesis, blood coagulation, and apoptosis [1Puente XS, Lopez-Otin C. A genomic analysis of rat protease and protease inhibitors Genome Res 2004; 14: 609-22.]. Accordingly, dysregulation of proteases have been implicated in numerous disease states such as cancer, osteoporosis, inflammatory disease, neurodegenerative disease, cardiovascular disease, type 2 diabetes, and acute injury [2Hooper NM. Proteases in biology and medicine. London: Portland Press 2002.]. Modulators to protease activities, either small molecules or antibodies, can be used as therapeutics to treat these diseases. Proteases constitute 5 to 10% of all pharmaceutical targets for small molecule drug discovery. To date, there have been six successful protease inhibitor drugs, including ACE inhibitors and HIV protease inhibitors [3Bogdanovic S, Langlands B. Proteases: Technologies and opportunities for drug discovery In: Westborough: D&MD Publications 2005.], with inhibitors for several additional proteases currently in clinical trials. With over 560 proteases or protease homolog coding regions annotated from the human genome [1Puente XS, Lopez-Otin C. A genomic analysis of rat protease and protease inhibitors Genome Res 2004; 14: 609-22.], this target class continues to be an important and active area for drug discovery.
Understanding protease function is critical to its application as a biomarker for physiological processes or for the development of disease treatments. The substrate specificity of proteases confers preferential targeting of its substrate in the presence of other peptides and proteins. Therefore, a better understanding of the binding to, and processing of, novel proteases with their natural substrates may help elucidate both their structure and function as well as suggest potential small molecule modulators [4Timmer JC, Salvesen GS. Caspase substrates Cell Death Differ 2007; 14: 66-72.]. Studying novel proteases using natural substrates can be technically difficult and therefore, many begin by first using short polypeptides as substrates. Standard methods of determining protease substrate specificity involve creating chemical combinatorial libraries of short polypeptides, typically tagged with fluorophores, and quencher molecule if the protease has a P' requirement [5Thormberry NA, Rano TA, Peterson EP, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B J Biol Chem 1997; 272: 17907-1.-7Choe Y, Leonetti F, Greenbaum DC, et al. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities J Biol Chem 2006; 281: 12824-32.]. This systematic approach provides a very rigorous and comprehensive method for determining substrate specificity. However it requires expertise and equipment for chemical synthesis, which are not readily available to most life science research laboratories. Fluorescent peptide substrates can also be purchased from commercial vendors but may be limited and costly. As an alternative to fluorescent peptides, some investigators have created protein fusion libraries which contain the potential substrate sequences and then rank specificity based upon the percent of cleaved product as measured by gel densitometry [8Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease Biochem Biophys Res Commun 2002; 294: 949-55.]. But this approach, in addition to being laborious and time consuming, has limited assay dynamic range and sensitivity.
Once the optimal peptide sequence has been established, most researchers use fluorescent peptides synthesized in-house or purchased from commercial sources, for their subsequent protease characterization studies. While adequate for many applications, these fluorescence assays may have limitations in sensitivity and dynamic range. Increasingly, investigators have turned to peptide conjugated aminoluciferin substrates. These bioluminescent substrates are significantly more sensitive and have wider dynamic ranges than analogous fluorescent substrates, typically 100 times more sensitive and 10-100 times wider dynamic range, in both biochemical and cell-based assay formats [9O'Brien MA, Daily WJ, Hesselberth E, et al. Homogeneous, bioluminescent protease assays: Caspase-3 as a model J Biomol Screen 2005; 10: 137-48.]. Additionally when performing a library screen for small molecule modulators, bioluminescent-based assays have been shown to be less hindered by non-specific compound interference than fluorescent-based assays [9O'Brien MA, Daily WJ, Hesselberth E, et al. Homogeneous, bioluminescent protease assays: Caspase-3 as a model J Biomol Screen 2005; 10: 137-48.-11Auld DS, Southall NT, Jadhav A, et al. Characterization of chemical libraries for luciferase inhibitory activity J Med Chem 2008; 51: 2372-86.].
We have previously described a novel biosensor using a genetically modified firefly luciferase that allows the facile interrogation of protease function without chemical synthesis [12Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51.]. It uses a bioluminescent substrate generated through molecular cloning and transcription/translation coupled cell-free expression. Thus protease substrates do not need to be purchased or chemically synthesized. The modified firefly luciferase is covalently joined at the native termini with a short peptide containing the protease recognition site which serves to restrict the luminescent reaction. Proteolytic cleavage of the peptide by the cognate protease activates the luciferase enzyme, typically over 100 fold. To express this mutant luciferase, new termini were inserted to create the circularly permuted form of firefly luciferase. The design strategy of this assay is shown in Fig. (1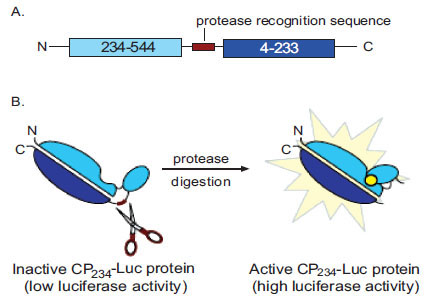 ). Importantly, we have shown that this mutant luciferase protease assay retains the advantages of the bioluminescent format, which include increased sensitivity and wide dynamic range, while accommodating proteases with and without P' requirements [12Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51.].
). Importantly, we have shown that this mutant luciferase protease assay retains the advantages of the bioluminescent format, which include increased sensitivity and wide dynamic range, while accommodating proteases with and without P' requirements [12Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51.].

Here we further demonstrate the utility of this mutant luciferase for protease detection, interrogation of multiple protease substrates, small molecule modulator screening, and inhibitor potency determination. For our model system, we inserted the Tobacco Etch Virus (TEV) protease recognition sequence into the circularly permuted firefly luciferase gene. Using standard molecular cloning techniques we generated twenty substrates, differing at the P1' position, and examined the TEV protease recognition sequence specificity. One of these substrates was then used to screen the Library of Pharmacologically Active Compounds (LOPAC) for small molecule inhibitors of TEV protease and determine the potency of a subset of these inhibitors.
MATERIALS AND METHODOLOGY
Primers were purchased from IDT. Sequencing was performed by Agencourt Bioscience Corporation. Unless otherwise specified, all other reagents are products of Promega Corporation (http://www.promega.com).
Evaluation of Different Sites for Circular Permutation
Four different structurally tolerant firefly luciferase sites were evaluated for insertion of the new mutant luciferase termini. The TEV protease recognition sequence with linkers (GSS-ENLYFQS-SSG) was inserted into the luc2 gene, a synthetic firefly (Photinus pyralis) luciferase, circularly permuted (CP) at Thr235, Glu269, Leu309, and Pro359. The constructs encoded fusion proteins of the following type: Met-(Luc2 residues Y-544)- GSS-ENLYFQS-SSG -(Luc2 residues 4-X)-Val (Table 1).
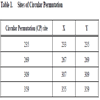
To synthesize these constructs, splice overlapping extension (SOE) PCR was performed [13CSH Protocols 2008.
[http://dx.doi.org/10.1101/pdb.prot4861] ]. Briefly, two general and two specific PCR primers for each construct were designed to generate two PCR products containing the two halves of the mutant luciferase and TEV protease recognition sequence. The PCR primers were designed such that the 3' end of the first PCR product (the first half of the mutant luciferase and TEV protease recognition sequence) overlapped with the 5' end of the second PCR product (the second half of the mutant luciferase and TEV protease recognition sequence). A second round of (primerless) PCR splices the two PCR fragments together. This is followed by a third and final round of PCR using the two external primers which results in a single full length product. The final PCR product was then gel purified and ligated into the Flexi vector pF9A. One positive clone each was sequence confirmed (Agencourt). These constructs were kindly supplied by B.F. Binkowski. The resultant clones were then transferred into the Flexi vector pF3K for expression in cell-free translation reactions. Transfer of the coding regions into Flexi vectors pF9A and pF3K were performed according to the manufacturer’s directions.
The two internal primers (2 and 3) were the same for all four constructs. Primer 2 was 5'-TTGGCACCGGAGCT CGATTGGAAGTACAGGTTCTCGCTCGAGCCCTTCTT GGCCTTAATGAGAATCTCGC -3' and primer 3 was 5'-AAGAAGGGCTCGAGCGAGAACCTGTACTTCCAATC GAGCTCCGGTGCCAAAAACATTAAGAAGGGCC -3'. The two unique PCR primers were as follows. For CP235, primer 1 was 5'- GCGATCGCCATGACCGCTATCCTCAG CGTGGTG -3' and primer 4 was 5'- GTTTAAACTCAGGG GATGATCTGGTTGCCGAAG -3'. For CP269, primer 1 was 5'- GCGATCGCCATGGAGGAGGAGCTATTCTTGC GCAG -3' and primer 4 was 5'- GTTTAAACTCAGCGGT ACATGAGCACGACCC -3'. For CP309, primer 1 was 5'- GCGATCGCCATGTTGCACGAGATCGCCAGCGG-3' and primer 4 was 5'- GTTTAAACTCAGCTTAGGTCGTA CTTGTCGATGAGAG-3'. For CP359, primer 1 was 5'- GCGATCGCCATGCCTGGCGCAGTAGGCAAGG-3' and primer 4 was 5'- GTTTAAACTCACCCTTCGGGGGTGAT CAGAATG-3'.
The first round of PCR reactions amplified the two halves of the mutant luciferase coding region in two independent reactions. PCR 1 reaction components were: 0.5 µL 50 ng/µL plasmid DNA template (containing the luc2 coding region); 5 µL 10X buffer; 5 µL 2 mM dNTPs; 2 µL 25 mM MgSO4; 33.5 µL dH2O; 1.5 µL 10 pmol/µL sense primer (primer 1 or 3); 1.5 µL 10 pmol/µL antisense primer (primer 2 or 4); 1 µL KOD Hot Start DNA Polymerase (Novagen). PCR 1 cycling parameters were: an initial denaturation step at 94°C for 2 minutes followed by 25 cycles of denaturation at 94°C for 30 seconds; annealing at 50°C for 30 seconds; extension at 68°C for 1 minute and a final extension of 68°C for 7 minutes. After the removal of the residual PCR primers using the Wizard SV Gel and PCR Clean-Up System, the second PCR reaction was performed. PCR 2 reaction components were: 1 ng/bp each purified PCR templates from the first round of PCR reactions; 5 µL 10X buffer; 5 µL 2 mM dNTPs; 2 µL 25 mM MgSO4; dH2O to 50 µL total volume; 1 µL KOD Hot Start DNA polymerase. PCR 2 cycling parameters were: an initial denaturation step at 94°C for 2 minutes followed by 10 cycles of denaturation at 94°C for 30 seconds; annealing at 55°C for 30 seconds; extension at 68°C for 1 minute and a final extension of 68°C for 7 minutes. PCR 3 immediately followed the PCR 2 reactions without template purification. The PCR 3 reaction components were: 1 µL PCR 2 product; 5 µL 10X buffer; 5 µL 2 mM dNTPs; 2 µL 25 mM MgSO4; 1.5 µL 10 pmol/µL outside primer 1; 1.5 µL 10 pmol/µL outside primer 4; 33 µL dH2O; 1 µL KOD Hot Start DNA polymerase. The PCR 3 cycling parameters were identical to the PCR 1 cycling parameters.
pF3K plasmid DNA encoding the circularly permuted luciferase with the TEV protease recognition sequence coding region, pF3K-CP235, 269, 309, 359-Luc/ENLYFQS, were isolated from E. coli using the PureYield Plasmid Midiprep kit and used as template in the TNT® SP6 High-Yield Protein Expression System according to the manufacturer’s instructions. Briefly, 2 µg of DNA (or water: “no DNA” lysate) was added to 30 µL master mix lysate plus 2 µL FluoroTect™ GreenLysin vitro Translation Labeling System for visualization on SDS-PAGE gels and the total volume was brought to 50 µL with dH2O. The 50 µL in vitro translation reactions were then incubated at 25°C for 2 hours. Fifteen µL of the reaction was then mixed with 15 µL of a 2X ProTEV protease buffer (100mM HEPES, 1mM EDTA, pH 7.0, plus fresh 2 mM DTT) with 1 µL of ProTEV protease (10U/µL) or dH2O. The digest reactions were incubated at 30°C for 30 minutes. Five µL of the digest reactions were placed in a white, flat bottom 96-well luminometer plate, in triplicate, and luminescence was measured following injection of 100 µL of Luciferase Assay Reagent using a Glomax® 96 microplate luminometer (3 second integration time). Five µL of the digest reactions were also size-fractionated on a 4-12% NuPAGE® SDS-PAGE gel (Invitrogen) in MES buffer and visualized on a fluorimager (ex. 488nm / em. 532nm, Typhoon® 9410, GE Healthcare Bio-Sciences).
P1' Specificity
To examine the P1' specificity of the TEV protease recognition sequence, we first created a vector with unique restriction sites. Here the luc2 gene was circularly permuted at Asp234, where X=233 and Y=234. The pF3A-based Flexi vector carries the CP234-Luc coding region with two unique restriction sites, NheI and BglII, inserted at the native luciferase N and C termini to facilitate the cloning in of protease recognition sequences. These two restriction sites also code for flexible linkers Ala-Ser and Gly-Ser on the N and C end of the protease recognition sequence, respectively. The glycine amino acid was created by destroying the BglII restriction site during the ligation step. Next, twenty oligonucleotide pairs were synthesized (IDT) which encoded for the polypeptide ENLYFQX, where X was all possible amino acids (Table 2). The oligonucleotides also contained the specific NheI and BglII 5' and 3' overhangs for direct ligation into the pF3A-CP234-Luc acceptor vector. The oligonucleotide pairs were hybridized together and ligation reactions performed using 50 ng linearized pF3A-CP234-Luc vector and 10 nM hybridized oligonucleotides according to manufacturer’s protocol. One positive clone each was then sequence confirmed (Agencourt).
CP234-Luc/ENLYFQX Protein Expression
The CP234-Luc/ENLYFQX plasmid DNA were isolated from E. coli using the PureYield Plasmid Midiprep kit and used as template in the TNT® SP6 High-Yield Protein Expression System according to the manufacturer’s instructions. Briefly, 2 µg of DNA (or water: “no DNA” lysate) was added to 30 µL master mix lysate plus 2 µL FluoroTect™ GreenLysin vitro Translation Labeling System for visualization on SDS-PAGE gels and the total volume was brought to 50 µL with dH2O. The 50 µL in vitro translation reactions were then incubated at 25°C for 2 hours. Fifteen µL of the reaction was then mixed with 15 µL of a 2X ProTEV protease buffer (100mM HEPES, 1mM EDTA, pH 7.0, plus fresh 2 mM DTT) with 1 µL of ProTEV protease (10U/µL) or dH2O. The digest reactions were incubated at 30°C for 30 minutes. Luminescence was measured from 5 µL of each digest reaction, in triplicate, following injection of 100 µL of Luciferase Assay Reagent using a MicroLumatPlus Microplate Luminometer LB96V (5 second integration time, EG&G Berthold). Five µL of the digest reactions were also size-fractionated on a 4-20% Criterion Tris-HCl SDS-PAGE gel (BioRad) in TGS buffer and visualized on a fluorimager (ex. 488nm / em. 532nm, Typhoon® 9410, GE Healthcare Bio-Sciences).
TEV Protease Limit of Detection and Linear Range using the CP234-Luc/ENLYFQS Assay
To determine the sensitivity and linearity of the CP234-Luc/ENLYFQS assay, titrating amounts of TEV protease were mixed with the CP234-Luc/ENLYFQS substrate (expressed using the TNT® SP6 High-Yield Protein Expression System) and luminescence was measured. Two µg CP234-Luc/ENLYFQS plasmid DNA (or water: “no DNA” lysate) was used as template and expressed as above. After expression, 15 µL of the lysate was mixed with 15 µL of a 2X ProTEV protease buffer plus titrating amounts of ProTEV protease (0 U to 3.16U). The digest reactions were incubated at 30°C for 30 minutes. Five µL of the digest reactions were placed in a white, flat bottom 96-well luminometer plate, in quadruplicate, and 100 µL of 1:1 diluted Bright-Glo™ Luciferase Assay Reagent in dH2O was added to each well. After incubation at room temperature for 5 minutes, luminescence was measured using a Glomax® 96 microplate luminometer (1 second integration time).
The signal to noise (S/N) was calculated from the luminescent values. S/N is defined as:
For this assay,
mean signal = mean signal RLU – the mean no-DNA RLU, at the various TEV concentrations
mean background = mean signal RLU – the mean no-DNA RLU, when no TEV enzyme was added
standard deviation of background = standard deviation of the signal RLU replicates, when no TEV enzyme was added
LOPAC1280 Library Screen
The Library of Pharmacologically Active Compounds (LOPAC1280; Sigma-Aldrich) was screened for TEV protease inhibitors in 384-well plates. CP234-Luc/ENLYFQC and a CP234-Luc/42 amino acid Gly/Ser rich peptide (CP234-Luc/42AA; previously described [12Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51.]) plasmid DNA were isolated from E. coli using the PureYield Plasmid Maxiprep kit. Six mL translation reactions for each were made using the TNT® SP6 High-Yield Protein Expression System. The reactions were gently rotated (~60 rpm) in a 50 mL conical tube for 2 hours at 25°C. The LOPAC1280 library of compounds was placed into two sets of four white, flat bottomed 384-well plates with each well containing 5 μL of compound at a concentration of 0.5 mM in DMSO. Columns 1, 2, 23, and 24 on each plate were used for solvent and enzyme control samples.
The library screens were performed using a Freedom EVO automation platform with a TeMo multi-channel pipetting system (Tecan). The translations were diluted into buffer as follows. For the CP234-Luc/ENLYFQC translation, 5.83 mL was added to 3.0 mL of 20X ProTEV protease buffer, 60 μL 1 M DTT and dH2O to a final volume of 35 mL. For the CP234-Luc/42AA, 5.83 mL was added to 3.0 mL 20X ProTEV protease buffer, 60 μL 1M DTT, 11.7 mL 10 mM HEPES, pH 7.5, and dH2O to 46.7 mL. To assay the effect of the compounds on the CP234-Luc/42AA, 20 μL of the CP234-Luc/42AA mixture was added to all wells in one set of four plates. To the second set of four plates, 5 μL ProTEV protease diluted to 0.5 U/μL in 10 mM HEPES, pH 7.5 was added, except for the no enzyme control lanes which received only dilution buffer. The same set of plates then received 15 μL of the CP234-Luc/ENLYFQC mix. The plates were spun down after each addition and were allowed to incubate 15 minutes at room temperature after the final addition. After this incubation, 25 μL Bright-Glo™ Luciferase Assay Reagent was added to each well of all plates. After incubation at room temperature for 5 minutes, the plates were read using a GENios Pro detection instrument (200 ms integration time; Tecan).
CP234-Luc/ENLYFQC Verification Assays
CP234-Luc/ENLYFQC verification assays were performed as follows. Fifteen µL TNT® SP6 High-Yield Protein Expression System lysate expressing CP234-Luc/ENLYFQC or CP234-Luc/42AA was mixed with 5 µL compound or vehicle, 7.5 U ProTEV protease and 1X ProTEV protease buffer (final concentration) in a total volume of 30 µL. The reactions were incubated at room temperature for 15 minutes and then stopped by adding 5 µL of the reaction, in triplicate, to 100 µL of a 1:1 dilution of Bright-Glo™ Luciferase Assay Reagent in dH2O. After 5 minutes at room temperature, luminescence was measured using a GloMax® 96 microplate luminometer (1 second integration time). TEV protease cleavage of CP234-Luc/ENLYFQC was normalized to CP234-Luc/42AA such that the ratio of CP234-Luc/ENLYFQC luminescence to CP234-Luc/42AA luminescence at no compound was 1.0. These normalized ratios were plotted against the logarithm of compound concentration and fit to a sigmoidal dose-response curve using GraphPad Prism 4 to estimate IC50 values.
Protein Fusion Cleavage Assays
A fusion protein of glutathione-S-transferase and maltose binding protein (GST-MBP) with the TEV protease recognition sequence and linkers (SGGGGG-ENLYFQA-IA) in-between the GST and MBP was created by cloning the MBP gene into the pFN2K Flexi vector. This vector carries the GST coding region upstream of the two unique restriction sites into which the MBP was cloned. The 5' unique restriction site, SgfI, codes for amino acids AIA, thus the first alanine becomes the P1' position for the TEV protease recognition sequence. This fusion protein was then expressed in KRX cells. The protein was purified using glutathione sepharose (GE Healthcare) as per the manufacturer’s directions. Protein concentration was estimated using Coomassie Plus Protein Assay Reagent (Pierce), using BSA as the standard.
One hundred µL reactions containing 35.5 µg GST-MBP, 1X ProTEV protease buffer, 1 mM DTT, titrating amounts of compound (or vehicle), and 10 U ProTEV protease were incubated at 30°C for 15 minutes. Reactions were stopped with the addition of 4X SDS sample buffer plus 200 mM DTT (final concentration was 1X SDS sample buffer plus 50 mM DTT). Samples were size fractionated on 4-20% Criterion Tris-HCl SDS-PAGE gels (BioRad) in TGS buffer. Gels were then stained with SimplyBlue SafeStain (Invitrogen) and destained overnight in water at room temperature. Gels were then scanned on a fluorimager (Typhoon® 9410, GE Healthcare Bio-Sciences) at 450V using no excitation or emission filters. Densitometry analysis (ImageQuant, GE Healthcare) was performed to determine percent cleavage of each sample. Each compound concentration was run in duplicate. Compounds were diluted in DMSO, except for NF-023 hydrate, which was diluted in dH2O. The above protein fusion cleavage assay conditions were designed such that the percent cleavage of the no compound control was 70-80%. Starting below 95-100% cleavage ensures the assay quantitatively measures inhibition. Percent cleavage data was plotted against the logarithm of compound concentration and fit to a sigmoidal dose-response curve using GraphPad Prism 4 to estimate the IC50 values.
RESULTS
Establishment of an Optimal CP Site for the Engineered Luciferase TEV Protease Assay
Four circularly permuted luciferases containing the TEV protease site were created: CP235, 269, 309, 359 -Luc/ENLYFQS. For each of these constructs, the site of circular permutation was chosen in a solvent exposed surface loop bounded by secondary structures such as a beta sheet or alpha helix using PDB file 1LCI (http://www.rcsb.org/pdb/home/home.do).
All four mutant luciferases were activated upon TEV protease digest as determined by both an increase in luminescence as well as the appearance on SDS-PAGE gel of the expected sized lower molecular weight CP-Luc protein fragments (Fig. 2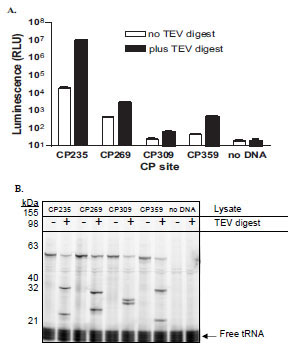 ). The four proteins were digested to a similar degree resulting in their predicted fragments: CP235-Luc, 34 and 26 kDa; CP269-Luc, 30.5 and 29.5 kDa; CP309-Luc, 25.5 and 34.5 kDa; and CP359-Luc, 20 and 40 kDa (Fig. 2B). The CP235-Luc/ENLYFQS protein generated significantly more light output, both before and after cleavage, than the other three proteins. In addition, protease digest of the CP235-Luc/ENLYFQS protein resulted in the largest fold activation (550 fold activation; Fig. 2A). In the case of the CP309-Luc/ENLYFQS and CP359-Luc/ENLYFQS, the undigested luminescent values were less than three standard deviations above the “no DNA” lysate values. This indicates that their basal luminescent values were within the background “noise” of the GloMax® 96 microplate luminometer and therefore fold activations could not be calculated. Based on these results, we chose to use a mutant luciferase circularly permuted near Thr235, at Asp234, for all our subsequent studies.
). The four proteins were digested to a similar degree resulting in their predicted fragments: CP235-Luc, 34 and 26 kDa; CP269-Luc, 30.5 and 29.5 kDa; CP309-Luc, 25.5 and 34.5 kDa; and CP359-Luc, 20 and 40 kDa (Fig. 2B). The CP235-Luc/ENLYFQS protein generated significantly more light output, both before and after cleavage, than the other three proteins. In addition, protease digest of the CP235-Luc/ENLYFQS protein resulted in the largest fold activation (550 fold activation; Fig. 2A). In the case of the CP309-Luc/ENLYFQS and CP359-Luc/ENLYFQS, the undigested luminescent values were less than three standard deviations above the “no DNA” lysate values. This indicates that their basal luminescent values were within the background “noise” of the GloMax® 96 microplate luminometer and therefore fold activations could not be calculated. Based on these results, we chose to use a mutant luciferase circularly permuted near Thr235, at Asp234, for all our subsequent studies.

Evaluation of TEV Protease Substrate Specificity
To enable both a comprehensive and easy method for interrogating protease specificity, we created a vector which carries the CP234 -Luc coding region with two unique restriction sites inserted at the native luciferase N and C termini to facilitate the cloning in of protease recognition sequences. Using this vector, we generated twenty bioluminescent protease substrates: CP234-Luc/ENLYFQX proteins with all possible amino acids at the P1' position, to assess the utility this assay for examining protease substrate specificity. Nineteen of the twenty were activated to various degrees upon TEV protease digest as determined by both an increase in luminescence (Fig. 3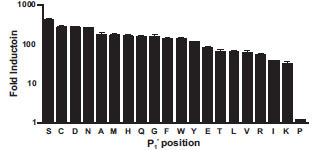 ) as well as the appearance of the smaller 34 kDa and 26 kDa CP234-Luc protein fragments (data not shown). The CP234-Luc/ENLYFQP protein was not activated by or digested by TEV protease. The fold activations of the proteins after protease digestion ranged from none (P1' = proline) to 420 (P1' = serine). Gel analysis of the digested luciferase fragment showed qualitatively similar results, although precise quantitation is difficult (data not shown). These results suggest that most amino acids are tolerated at the P1' position, with the exception of proline. This is consistent with previous reports [8Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease Biochem Biophys Res Commun 2002; 294: 949-55.].
) as well as the appearance of the smaller 34 kDa and 26 kDa CP234-Luc protein fragments (data not shown). The CP234-Luc/ENLYFQP protein was not activated by or digested by TEV protease. The fold activations of the proteins after protease digestion ranged from none (P1' = proline) to 420 (P1' = serine). Gel analysis of the digested luciferase fragment showed qualitatively similar results, although precise quantitation is difficult (data not shown). These results suggest that most amino acids are tolerated at the P1' position, with the exception of proline. This is consistent with previous reports [8Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease Biochem Biophys Res Commun 2002; 294: 949-55.].

Determination of TEV Protease Substrate Detection Limit and Linear Range
To determine the sensitivity and linear range of the CP234-Luc/ENLYFQS assay, a TEV enzyme titration was performed. The luminescence from the CP234-Luc/ENL-YFQS assay was dependent on the TEV enzyme concentration. The signal to noise (S/N) was calculated from the luminescent values and is defined as the mean signal – mean background divided by the standard deviation of the background (Fig. 4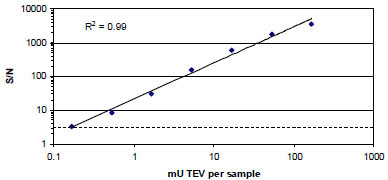 ). The limit of detection was defined as the amount of TEV protease detected at a S/N of 3 (i.e., 3 standard deviations higher than the background). Thus the limit of detection was 0.163 mU TEV protease per 100 μL Bright GloTM Luciferase Assay sample. The linear range of the assay was ≥ 1,000 fold (0.163 to 167 mU (the highest concentration tested in this experiment) of TEV protease per 100 μL Bright GloTM Luciferase Assay sample).
). The limit of detection was defined as the amount of TEV protease detected at a S/N of 3 (i.e., 3 standard deviations higher than the background). Thus the limit of detection was 0.163 mU TEV protease per 100 μL Bright GloTM Luciferase Assay sample. The linear range of the assay was ≥ 1,000 fold (0.163 to 167 mU (the highest concentration tested in this experiment) of TEV protease per 100 μL Bright GloTM Luciferase Assay sample).

Screening of TEV Protease Inhibitors
TEV protease is widely used as a tool to cleave an affinity tag from the protein of interest after protein purification. Therefore, it is desirable to have an inhibitor that can “turn off” TEV protease after fusion protein cleavage, but does not affect subsequent processing steps. Our initial attempts using known cysteine inhibitors either failed to inhibit TEV protease or affected downstream applications. To find a suitable inhibitor, we screened the LOPAC1280 library compounds in 384-well format at 100 μM using the CP234-Luc/ENLYFQC protease assay to identify potential small-molecule inhibitors.
To control for non-specific CP234-Luc affecters, we used a previously described CP234-Luc protein containing a 42 amino acid glycine/serine rich sequence [12Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51.]. This 42 amino acid glycine/serine rich sequence is not recognized by native proteases in the TnT® SP6 High-Yield lysate or by the TEV protease (data not shown). Because of the long length of this polypeptide, the basal luminescence of the CP234-Luc/42AA protein was approximately 10 times greater than the luminescence of the CP234-Luc/ENLYFQC protein after TEV protease digestion. The luminescence of the CP234-Luc/42AA protein was determined by incubating the protein in the TEV assay conditions with DMSO solvent. The luminescence of the CP234-Luc/42AA protein was also measured in the presence of the LOPAC1280 library. Therefore, non-specific affecters were detected by a change in the CP234-Luc/42AA protein luminescent signal from the DMSO solvent control. Incubation with twenty-five LOPAC1280 library compounds (out of 1280 compounds, 2%) resulted in a CP234-Luc/42AA luminescent signal outside three standard deviations from the mean of the DMSO solvent control. Presumably these compounds either interfere with CP234-Luc activity or, as with highly colored compounds, they may be quenching the luminescent signal produced by the CP234-Luc [11Auld DS, Southall NT, Jadhav A, et al. Characterization of chemical libraries for luciferase inhibitory activity J Med Chem 2008; 51: 2372-86.].
Hits were defined as those compounds which inhibited TEV protease activity greater than 60%. We chose to only examine these compounds because our goal was to find a very potent inhibitor of TEV protease activity. Representative screen data from plate 1 is shown (Fig. 5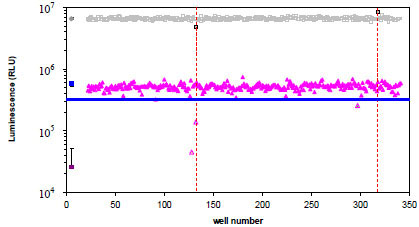 ). On plate 1 there were three hits based on greater than 60% TEV protease inhibition: one was confirmed as an inhibitor, aurintricarboxylic acid (ATA; yellow triangle), one was not re-tested (green triangle) and one also caused a reduction in CP234-Luc/42AA assay activity (blue triangle and gray/black square), suggesting that the compound is non-specifically affecting the luminescent reaction. One additional compound (gray/black square) caused an increase in CP234-Luc/42AA activity; however it did not appear to affect the CP234-Luc/ENLYFQC assay activity.
). On plate 1 there were three hits based on greater than 60% TEV protease inhibition: one was confirmed as an inhibitor, aurintricarboxylic acid (ATA; yellow triangle), one was not re-tested (green triangle) and one also caused a reduction in CP234-Luc/42AA assay activity (blue triangle and gray/black square), suggesting that the compound is non-specifically affecting the luminescent reaction. One additional compound (gray/black square) caused an increase in CP234-Luc/42AA activity; however it did not appear to affect the CP234-Luc/ENLYFQC assay activity.

 ) based on greater than 60% TEV protease inhibition, blue line (= 345,311 RLU). The CP234-Luc/42AA assay was also performed to detect potential non-specific affectors of the CP234-Luc assay and the results, which are overlaid on the graph in gray, indicate that two compounds non-specifically affected the assay. (
) based on greater than 60% TEV protease inhibition, blue line (= 345,311 RLU). The CP234-Luc/42AA assay was also performed to detect potential non-specific affectors of the CP234-Luc assay and the results, which are overlaid on the graph in gray, indicate that two compounds non-specifically affected the assay. ( ) = CP234-Luc/42AA activity in DMSO; (
) = CP234-Luc/42AA activity in DMSO; ( ) = CP234-Luc/42AA activity with compound library; (
) = CP234-Luc/42AA activity with compound library; ( ) = CP234-Luc/ENLYFQC activity minus TEV enzyme in DMSO; (
) = CP234-Luc/ENLYFQC activity minus TEV enzyme in DMSO; ( ) = CP234-Luc/ENLYFQC activity plus TEV enzyme in DMSO, and (
) = CP234-Luc/ENLYFQC activity plus TEV enzyme in DMSO, and ( ) = CP234-Luc/ENLYFQC activity plus TEV protease with compound library. Controls are plotted as the mean of N = 64 (CP234-Luc/42AA in DMSO) or 32 (CP234-Luc/ENLYFQC ± TEV protease in DMSO). Error bars are the standard deviation of the mean.
) = CP234-Luc/ENLYFQC activity plus TEV protease with compound library. Controls are plotted as the mean of N = 64 (CP234-Luc/42AA in DMSO) or 32 (CP234-Luc/ENLYFQC ± TEV protease in DMSO). Error bars are the standard deviation of the mean.In total, twenty-one compounds inhibited greater than 60% of the TEV protease cleavage of CP234-Luc/ENLYFQC (out of 1280 compounds, 1.6%). Nine of the 21 compounds which inhibited greater than 60% of the TEV protease cleavage of CP234-Luc/ENLYFQC also resulted in a CP234-Luc/42AA luminescent signal outside three standard deviations from the mean of the DMSO solvent control and thus were excluded from further analysis. The remaining 12 compounds were, therefore, potential TEV protease specific inhibitors (out of 1280 compounds, 0.9%).
Five of the 12 compounds were re-tested using both the CP234-Luc/ENLYFQC assay and the protein fusion cleavage assay. We did not test all twelve compounds because of cost considerations. Instead, we chose to re-test a range of potential TEV protease inhibitors: three potent inhibitors (inhibited >80% cleavage activity) and two moderate inhibitors (inhibited 60-70% cleavage activity). The five compounds were: aurintricarboxylic acid (ATA), 8,8'-[carbonylbis (imino-3,1-phenylenecarbonylimino)]bis(1,3,5-naphthalene-trisulfonic acid) hexasodium salt (NF-023), 4-[3-(4-Acetyl-3-hydroxy-2-propylphenoxy)propoxy]phenoxyacetic acid (L-165,041), SCH-202676 hydrobromide or N-(2,3-diphenyl-1,2,4-thiadiazol-5-(2H)-ylidene)methanamine hydrobromide (SCH HBr), and 6-hydroxyl-DL-DOPA or 2,5-dihydroxy-DL-tyrosine (L-DOPA). Three out of the five compounds showed dosage-dependent inhibition as shown in Fig. (6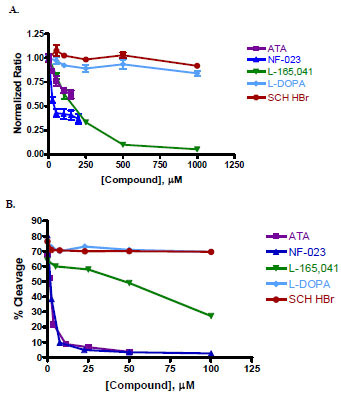 ). For the CP234-Luc/ENLYFQC assay, NF-023 and ATA were tested from 0 – 0.2 mM and the other three compounds were tested from 0 – 1 mM (Fig. 6A). For the protein fusion cleavage assay, compounds were tested from 0 – 0.1 mM (Fig. 6B). Inhibitor potency ranking was determined by estimating the IC50 values (see Materials and Methodology; Table 3).
). For the CP234-Luc/ENLYFQC assay, NF-023 and ATA were tested from 0 – 0.2 mM and the other three compounds were tested from 0 – 1 mM (Fig. 6A). For the protein fusion cleavage assay, compounds were tested from 0 – 0.1 mM (Fig. 6B). Inhibitor potency ranking was determined by estimating the IC50 values (see Materials and Methodology; Table 3).


Of the five compounds, SCH HBr and L-DOPA did not inhibit TEV protease cleavage in either assay re-test. The potency ranking using the two assays was the same for two out of the three remaining compounds. In the case of ATA, an IC50 value could not be determined using the CP234-Luc/ENLYFQC assay. This was most likely due to assay interference by the dark red color of ATA. ATA (by protein fusion cleavage assay) and NF-023 were determined to be potent inhibitors of TEV protease cleavage while L-165,041 was a slightly weaker inhibitor.
DISCUSSION
We have previously described the general applicability of a protease assay for substrate identification, with and without P' requirements, using a genetically engineered firefly luciferase biosensor, which is highly sensitive and has a wide dynamic range (Fig. 1) [12Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51.]. Furthermore, this assay does not require the purchase of, or chemical synthesis of, peptide substrates. In this report, we further demonstrate the utility of this assay for protease detection, substrate specificity analysis, small molecule inhibitor screening, and potency determination using TEV protease as a model system.
The TEV protease used in this study (ProTEV protease) is a 50kDa version of the NIa protease from TEV that has been engineered to be more stable than native TEV protease [8Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease Biochem Biophys Res Commun 2002; 294: 949-55., 14Dougherty WG, Parks TD. Post-translational processing of the tobacco etch virus 49-kDa small nuclear inclusion polyprotein: Identification of an internal cleavage site and delimitation of VPg and proteinase domains Virology 1991; 183: 449-56., 15Carrington JC, Haldman R, Dolja VV, Restrepo-Hartwig MA. Internal cleavage and trans-proteolytic activities of the VPg-proteinase (NIa) of tobacco etch potyvirus in vivo J Virol 1993; 67: 6995-7000.]. TEV protease is a highly site-specific cysteine protease that recognizes the seven amino acid sequence EXXYXQ(G/S). This site is most commonly ENLYFQG, with cleavage occurring between glutamine and glycine or serine [16Dougherty WG, Parks TD, Cary SM, Bazan JF, Fletterick RJ. Characterization of the catalytic residues of the tobacco etch virus 49-kDa proteinase Virology 1989; 172: 302-10., 17Carrington JC, Dougherty WG. A viral cleavage site cassette: identification of amino acid sequences required for tobacco etch virus polyprotein processing Proc Natl Acad Sci USA 1988; 85: 3391-5.]. However, the protease will recognize and cleave sequences with a variety of amino acids at the G/S (or P1') position [8Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease Biochem Biophys Res Commun 2002; 294: 949-55.]. The protease is used primarily to cleave affinity tags from fusion proteins after protein purification.
We have previously optimized the linker length and site of circular permutation for a non-lytic, live cell cAMP biosensor which resulted in dramatic performance improvements [12Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51.]. However, our previous protease assay optimizations were only focused on the length of the polypeptide inserted between the native N and C termini [12Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51.]. In this study, we examined the performance of the CP-Luc/ENLYFQS proteins circularly permuted at different sites (Fig. 2). All four proteins were similarly digested by the TEV protease (Fig. 2B). Therefore to determine the optimal circularly permuted site, we compared the luminescent values and fold activations of the four proteins (Fig. 2A). To calculate fold activation, it is necessary that the basal luminescent values are greater than three standard deviations above the “no DNA” control. CP309-Luc/ENLYFQS and CP359-Luc/ENLYFQS basal luminescent values were within three standard deviations of the “no DNA control” and, since we could not calculate their fold activations, they were not chosen. A comparison of the luminescent values and fold activations of the remaining two proteins, CP235 -Luc/ENLYFQS and CP269 -Luc/ENLYFQS, showed that the most optimal protein in terms of basal and activated luminescence as well as fold activation was the CP235 -Luc/ENLYFQS protein (Fig. 2A). We therefore used a mutant luciferase circularly permuted near Thr235, at Asp234, for all our subsequent studies (Figs. 3-6).
One of our main objectives for this protease assay was to facilitate the easy and rapid evaluation of multiple protease substrates. Although other methodologies exist, they can be prohibitive in terms of the cost to purchase the substrates or in the expertise required to synthesize the substrates. In the case of protein fusion cleavage assays, they are laborious to perform and are not well suited for high throughput screening applications. We therefore devised a bioluminescent-based CP234-Luc cloning method where substrates could be quickly and easily generated. Using this method, we generated 20 substrates and performed a TEV protease substrate specificity study. Our results are consistent with previous reports [8Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease Biochem Biophys Res Commun 2002; 294: 949-55.]. Furthermore, the dynamic range for the CP234 -Luc/ENLYFQS P1' substrate was 420 fold (Fig. 3).
One limitation of the CP234 -Luc method is that rigorous enzymatic analyses are difficult to perform. Since the protease substrates are generated in a cell-free lysate, the exact amount of substrate is hard to determine. Consequently, exact quantitation analysis is challenging. Other methodologies also have limitations quantitatively measuring protease activity. For example, it is important that the protein fusion cleavage assay conditions are engineered such that the substrate digestions are not allowed to reach 100% completion. If not, multiple substrates may appear to be equally preferred by the protease. This was demonstrated by the near complete intracellular processing of multiple substrates by TEV protease in E. coli [8Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease Biochem Biophys Res Commun 2002; 294: 949-55.]. Another potential limitation of protein fusion cleavage assays is their dependence on gel conditions and the densitometer dynamic range (sample loading, scan settings, etc.).
Nevertheless, the results from our semi-quantitative analysis based on fold of activation (Fig. 3) are consistent with results using other methods in previous reports [8Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease Biochem Biophys Res Commun 2002; 294: 949-55.]. The relative ranking of ten amino acids at the P1' position between the CP234 -Luc/ENLYFQS assay and an in vitro processing (protein fusion cleavage) assay were similar with two exceptions: Asp and Gln. The relative ranking of those two amino acids at the P1' position was significantly higher using CP234 -Luc/ENLYFQS assay than the in vitro processing gel cleavage assay [8Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease Biochem Biophys Res Commun 2002; 294: 949-55.]. One possible explanation for this difference may be the context in which the protease recognition sequence is presented. Since the context is non-native in both assays, it is possible that the differences observed in the relative rankings are due to the ability of the protease to access its substrate.
The CP234 -Luc/ENLYFQS assay, similar to what we have previously demonstrated using a caspase-3 substrate (CP234 -Luc/DEVDG) [12Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51.], has a wide dynamic range, ≥1,000 fold, and is highly sensitive with a limit of detection of 0.163 mU TEV protease / sample (Fig. 4). Unfortunately, we cannot compare the sensitivity of the CP234 -Luc/ENLYFQS assay with an analogous fluorescent assay because, to the best of our knowledge, such an assay is not commercially available. However, we can compare the CP234 -Luc/DEVDG assay [12Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51.] with both analogous fluorescent assays as well as an analogous peptide conjugated aminoluciferin protease substrate assay [9O'Brien MA, Daily WJ, Hesselberth E, et al. Homogeneous, bioluminescent protease assays: Caspase-3 as a model J Biomol Screen 2005; 10: 137-48.]. The comparison between the assays can be estimated after normalizing the two different sources of caspase-3 protease (and assuming that the substrates used to determine specific activity are comparable). We estimated that the sensitivity of the CP234-Luc/DEVDG assay was 10 to 100 fold more sensitive than the two analogous fluorescent substrates tested; Rho110 and AMC and ~10 fold less sensitive than the Z-DEVD-aminoluciferin protease substrate assay. These results demonstrate that the CP234 -Luc assay retains the high sensitivity and wide dynamic range afforded bioluminescence-based assays while enabling researchers to easily generate protease substrates of interest.
TEV protease is widely used as a tool to cleave a protein purification tag from the protein of interest. Therefore, we wanted to find an inhibitor which could “turn off” TEV protease after fusion protein cleavage, but did not affect subsequent processing steps. Initially, we tried several established inhibitors without success. For example, while 5 mM zinc effectively inhibited the protease, it also frequently precipitated the proteins out of solution. Similarly, inactivation of TEV, a cysteine protease, with sulfhydryl modifying reagents such as N-ethylmaleimide and iodoacetamide required concentrations at levels where other proteins in the solution were also modified. And finally, TEV protease proved resistant to various known inhibitors of cysteine proteases, including E-64 protease inhibitor, ALLN (N-acetyl-Leu-Leu-NIe-CHO), and EST ((2S,3S)-trans-Epoxysuccinyl-L-leucylamido-3-methylbutane Ethyl Ester) (data not shown). A peptide substrate analog with a chloromethylketone (CMK) in the P1' position was synthesized according to previous studies [18Shaw E, Mares-Guia M, Cohen W. Evidence for an active-center histidine in trypsin through use of a specific reagent 1-chloro-3-tosylamido-7-amino-2-heptanona, the chloromethyl ketone derived from N-α-tosyl-l-lysine Biochemistry 1965; 4: 2219-4.]. This hexapeptide peptide CMK inhibitor was able to inhibit TEV protease cleavage with an IC50 of ~5.7 µM as measured by the protein fusion cleavage assay. Complete inhibition was observed at 20 µM after a 15 minute pre-incubation with the protease before adding substrate (data not shown). However, the cost of the peptide-based inhibitor prohibited its practical use.
We screened the LOPAC1280 library in 384-well format to find a small molecule inhibitor of TEV protease which would not affect subsequent processing steps. The library was screened at ten times the concentration of a typical screen (100 µM versus 10 µM) (Fig. 5). We chose to perform the screen at this high concentration to increase the probability of finding a very potent inhibitor of TEV protease. The high compound concentration likely resulted in a higher than expected number [9O'Brien MA, Daily WJ, Hesselberth E, et al. Homogeneous, bioluminescent protease assays: Caspase-3 as a model J Biomol Screen 2005; 10: 137-48.-11Auld DS, Southall NT, Jadhav A, et al. Characterization of chemical libraries for luciferase inhibitory activity J Med Chem 2008; 51: 2372-86.] of non-specific affecters of CP234-Luc/42AA luminescence, in part due to increased color quenching. Of the 25 compounds which non-specifically affected the CP234-Luc/42AA luminescent signal, nine were visibly colored at 100 µM, four have been previously shown to inhibit luciferase activity (M.A. O’Brien, personal communication); three appeared to increase activity and the remaining nine compounds reduced activity. It would be interesting to determine the number of compounds which affect the CP234-Luc/42AA luminescent signal when screened at 10 µM concentration.
Five of the 12 compound hits found in the primary screen were re-tested using the same CP234-Luc/ENLYFQC assay as well as a protein fusion cleavage assay. Protein fusion cleavage is the most commonly used assay to assess inhibition of TEV protease activity. The results from the two assays were the same in terms of confirming the three hits as well as the relative ranking of two of the three hits (Fig. 6; Table 3). The estimated IC50 for L-165,041 was similar (within 10 fold) between the two assays, but the estimated IC50 for NF-023 was significantly different between the two assays. Although the reason for this difference is not understood, it is well known that different assays and conditions can result in different IC50 values. It is also worth noting that the P1' position of the TEV protease recognition sequence was not the same between the CP234-Luc/ENLYFQC and protein fusion cleavage assays, cysteine versus alanine, respectively. The experiments for which the TEV protease inhibitor was intended required that the P1' amino acid be a cysteine. And since all bioluminescent substrates were readily available, we were able to choose that substrate for the screening study. But for the protein fusion cleavage assay, we did not have the option of choosing the P1' amino acid since alanine was pre-engineered into the pFN2K parent vector. Fortunately in this case, based on the P1' position study (Fig. 3), we do not believe that the variations observed in the NF-023 IC50 values were a result of this difference. In other applications, however, the ability to precisely choose the exact protease substrate may be critical.
The three confirmed TEV protease inhibitors were: aurintricarboxylic acid (ATA), 8,8'-[carbonylbis(imino-3,1-phenylenecarbonylimino)]bis(1,3,5-naphthalene-trisulfonic acid) hexasodium salt (NF-023), and 4-[3-(4-Acetyl-3-hydroxy-2-propylphenoxy)propoxy]phenoxyacetic acid (L-165,041). The chemical structures of the three compounds are shown in Fig. (7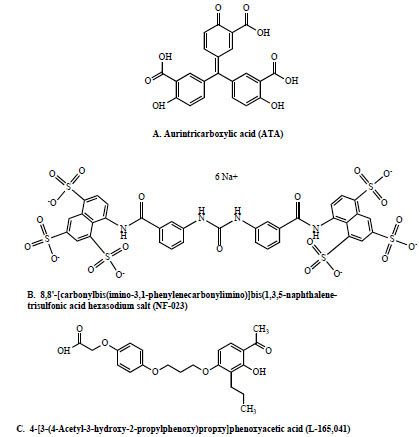 ). ATA and NF-023 were strong TEV protease inhibitors while L-165,041 was a moderate inhibitor. Although ATA is listed as a DNA topoisomerase II inhibitor in the Sigma LOPAC library database, it has also been shown to inhibit cysteine proteases such as calpain [19Posner A, Raser KJ, Hajimohammadreza I, Yuen PW, Wang KK. Aurintricarboxylic acid is an inhibitor of mu- and m-calpain Biochem Mol Biol Int 1995; 36: 291-9.] and caspases 3, 6, 7, and 9 [20Mesner Jr PW, Bible KC, Martins LM, et al. Characterization of caspase processing and activation in HL-60 cell cytosol under cell-free conditions. Nucleotide requirement and inhibitor profile J Biol Chem 1999; 274: 22635-645.]. Therefore, our results showing an inhibitory effect of ATA on the TEV cysteine protease are consistent with previous reports. NF-023, listed as a potent, selective P2X1 receptor antagonist in the Sigma LOPAC library database, is an analog of suramin, a hexasulfonated naphthylurea. Suramin has many, widely diverse uses such as to treat trypanosomiasis and onchocerciasis [21Hawking F. Suramin: with special reference to onchocerciasis Adv Pharmacol Chemother 1978; 15: 289-322.], as an anti-tumor drug [22Stein CA, LaRocca RV, Thomas R, McAtee N, Myers CE. Suramin: an anticancer drug with a unique mechanism of action J Clin Oncol 1989; 7: 499-508.-25La Rocca RV, Stein CA, Danesi R, Cooper MR, Uhrich M, Myers CE. A pilot study of suramin in the treatment of metastatic renal cell carcinoma Cancer 1991; 67: 1509-3.], Malaria parasite Plamodium falciparum erythrocyte invasion inhibitor [26Fleck SL, Birdsall B, Babon J, et al. Suramin and suramin analogues inhibit merozoite surface protein-1 secondary processing and erythrocyte invasion by the malaria parasite Plasmodium falciparum J Biol Chem 2003; 278: 47670-77.], HIV-1 reverse transcriptase inhibitor [27Jentsch KD, Hunsmann HH, Nickel P. Inhibition of human immunodeficiency virus type I reverse transcriptase by suramin-related compounds J Gen Virol 1987; 68: 2183-92.], and inhibitor of three neutrophil serine proteinases: neutrophil elastase, cathepsin G and proteinase 3 [28Cadène M, Duranton J, North A, Si-Tahar M, Chignard M, Bieth JG. Inhibition of neutrophil serine proteinases by suramin J Biol Chem 1997; 272: 9950-55.]. Interestingly, TEV protease is a serine-like cysteine protease. Thus it is possible that NF-023’s inhibitory mechanism of action on TEV protease may be the same as suramin’s on the neutrophil serine proteinases. Further supporting this idea is the fact that two (out of 12) additional hits found in the primary LOPAC screen were suramin and NF-449, another suramin analog. This suggests that a structure-activity relationship exists between suramin and related compounds and their TEV protease inhibitory properties. Finally, L-165,041 is a peroxisome proliferator-activated receptor beta agonist. A search of the literature failed to yield any previous reports which would help explain this compound’s TEV protease inhibitory effects.
). ATA and NF-023 were strong TEV protease inhibitors while L-165,041 was a moderate inhibitor. Although ATA is listed as a DNA topoisomerase II inhibitor in the Sigma LOPAC library database, it has also been shown to inhibit cysteine proteases such as calpain [19Posner A, Raser KJ, Hajimohammadreza I, Yuen PW, Wang KK. Aurintricarboxylic acid is an inhibitor of mu- and m-calpain Biochem Mol Biol Int 1995; 36: 291-9.] and caspases 3, 6, 7, and 9 [20Mesner Jr PW, Bible KC, Martins LM, et al. Characterization of caspase processing and activation in HL-60 cell cytosol under cell-free conditions. Nucleotide requirement and inhibitor profile J Biol Chem 1999; 274: 22635-645.]. Therefore, our results showing an inhibitory effect of ATA on the TEV cysteine protease are consistent with previous reports. NF-023, listed as a potent, selective P2X1 receptor antagonist in the Sigma LOPAC library database, is an analog of suramin, a hexasulfonated naphthylurea. Suramin has many, widely diverse uses such as to treat trypanosomiasis and onchocerciasis [21Hawking F. Suramin: with special reference to onchocerciasis Adv Pharmacol Chemother 1978; 15: 289-322.], as an anti-tumor drug [22Stein CA, LaRocca RV, Thomas R, McAtee N, Myers CE. Suramin: an anticancer drug with a unique mechanism of action J Clin Oncol 1989; 7: 499-508.-25La Rocca RV, Stein CA, Danesi R, Cooper MR, Uhrich M, Myers CE. A pilot study of suramin in the treatment of metastatic renal cell carcinoma Cancer 1991; 67: 1509-3.], Malaria parasite Plamodium falciparum erythrocyte invasion inhibitor [26Fleck SL, Birdsall B, Babon J, et al. Suramin and suramin analogues inhibit merozoite surface protein-1 secondary processing and erythrocyte invasion by the malaria parasite Plasmodium falciparum J Biol Chem 2003; 278: 47670-77.], HIV-1 reverse transcriptase inhibitor [27Jentsch KD, Hunsmann HH, Nickel P. Inhibition of human immunodeficiency virus type I reverse transcriptase by suramin-related compounds J Gen Virol 1987; 68: 2183-92.], and inhibitor of three neutrophil serine proteinases: neutrophil elastase, cathepsin G and proteinase 3 [28Cadène M, Duranton J, North A, Si-Tahar M, Chignard M, Bieth JG. Inhibition of neutrophil serine proteinases by suramin J Biol Chem 1997; 272: 9950-55.]. Interestingly, TEV protease is a serine-like cysteine protease. Thus it is possible that NF-023’s inhibitory mechanism of action on TEV protease may be the same as suramin’s on the neutrophil serine proteinases. Further supporting this idea is the fact that two (out of 12) additional hits found in the primary LOPAC screen were suramin and NF-449, another suramin analog. This suggests that a structure-activity relationship exists between suramin and related compounds and their TEV protease inhibitory properties. Finally, L-165,041 is a peroxisome proliferator-activated receptor beta agonist. A search of the literature failed to yield any previous reports which would help explain this compound’s TEV protease inhibitory effects.

The LOPAC1280 library screen for TEV protease inhibitors demonstrates that the CP234-Luc protease assay can be used to screen for small molecule modulators in a high throughput 384-well format. More studies will be needed to fully validate its utility for high throughput screening, for example, improving assay quality (i.e., Z’ and Z values currently around 0.4-0.5) on a larger and more diverse compound library. Nevertheless, we were able to identify several novel TEV protease inhibitors. The use of these inhibitors to “turn off” TEV protease for applications in protein purification is being explored.
CONCLUSION
Proteases play a critical role in almost every aspect of biology. Therefore a better understanding of their often times complex functions and regulation, is vital to disease prevention and treatment. One of the first steps when investigating novel proteases are to establish its substrate specificity and a robust assay. Here we have demonstrated the utility of a genetically engineered luciferase protease assay which allows for the rapid and sensitive evaluation of multiple substrates without the need for chemically synthesize peptide substrates, made in-house or purchased. For many, this will enable a more comprehensive study of novel proteases with the added increase in sensitivity and dynamic range afforded by the bioluminescent-based format. Once an optimal substrate sequence has been determined, that substrate can be directly used for applications such as protease detection, high throughput screening for protease modulators, and their further characterizations.
ACKNOWLEDGEMENTS
We would like to thank Brock Binkowski, Braeden Butler, Susan Frackman, Erika Hawkins, Martha O’Brien, Ethan Strauss and Monica Wood for insightful discussions and contributions of technical expertise.
REFERENCES
| [1] | Puente XS, Lopez-Otin C. A genomic analysis of rat protease and protease inhibitors Genome Res 2004; 14: 609-22. |
| [2] | Hooper NM. Proteases in biology and medicine. London: Portland Press 2002. |
| [3] | Bogdanovic S, Langlands B. Proteases: Technologies and opportunities for drug discovery In: Westborough: D&MD Publications 2005. |
| [4] | Timmer JC, Salvesen GS. Caspase substrates Cell Death Differ 2007; 14: 66-72. |
| [5] | Thormberry NA, Rano TA, Peterson EP, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B J Biol Chem 1997; 272: 17907-1. |
| [6] | Harris JL, Niles A, Burdick K, et al. Definition of the extended substrate specificity determinants for β-Tryptases I and II J Biol Chem 2001; 276: 34941-7. |
| [7] | Choe Y, Leonetti F, Greenbaum DC, et al. Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities J Biol Chem 2006; 281: 12824-32. |
| [8] | Kapust RB, Tözsér J, Copeland TD, Waugh DS. The P1' specificity of tobacco etch virus protease Biochem Biophys Res Commun 2002; 294: 949-55. |
| [9] | O'Brien MA, Daily WJ, Hesselberth E, et al. Homogeneous, bioluminescent protease assays: Caspase-3 as a model J Biomol Screen 2005; 10: 137-48. |
| [10] | Valley MP, Zhou W, Hawkins EM, et al. A bioluminescent assay for monoamine oxidase activity Anal Biochem 2006; 359: 238-46. |
| [11] | Auld DS, Southall NT, Jadhav A, et al. Characterization of chemical libraries for luciferase inhibitory activity J Med Chem 2008; 51: 2372-86. |
| [12] | Fan F, Binkowski B, Butler B, Stecha P, Lewis K, Wood K. Novel genetically encoded biosensors using firefly luciferase ACS Chem Biol 2008; 3: 346-51. |
| [13] | CSH Protocols 2008. [http://dx.doi.org/10.1101/pdb.prot4861] |
| [14] | Dougherty WG, Parks TD. Post-translational processing of the tobacco etch virus 49-kDa small nuclear inclusion polyprotein: Identification of an internal cleavage site and delimitation of VPg and proteinase domains Virology 1991; 183: 449-56. |
| [15] | Carrington JC, Haldman R, Dolja VV, Restrepo-Hartwig MA. Internal cleavage and trans-proteolytic activities of the VPg-proteinase (NIa) of tobacco etch potyvirus in vivo J Virol 1993; 67: 6995-7000. |
| [16] | Dougherty WG, Parks TD, Cary SM, Bazan JF, Fletterick RJ. Characterization of the catalytic residues of the tobacco etch virus 49-kDa proteinase Virology 1989; 172: 302-10. |
| [17] | Carrington JC, Dougherty WG. A viral cleavage site cassette: identification of amino acid sequences required for tobacco etch virus polyprotein processing Proc Natl Acad Sci USA 1988; 85: 3391-5. |
| [18] | Shaw E, Mares-Guia M, Cohen W. Evidence for an active-center histidine in trypsin through use of a specific reagent 1-chloro-3-tosylamido-7-amino-2-heptanona, the chloromethyl ketone derived from N-α-tosyl-l-lysine Biochemistry 1965; 4: 2219-4. |
| [19] | Posner A, Raser KJ, Hajimohammadreza I, Yuen PW, Wang KK. Aurintricarboxylic acid is an inhibitor of mu- and m-calpain Biochem Mol Biol Int 1995; 36: 291-9. |
| [20] | Mesner Jr PW, Bible KC, Martins LM, et al. Characterization of caspase processing and activation in HL-60 cell cytosol under cell-free conditions. Nucleotide requirement and inhibitor profile J Biol Chem 1999; 274: 22635-645. |
| [21] | Hawking F. Suramin: with special reference to onchocerciasis Adv Pharmacol Chemother 1978; 15: 289-322. |
| [22] | Stein CA, LaRocca RV, Thomas R, McAtee N, Myers CE. Suramin: an anticancer drug with a unique mechanism of action J Clin Oncol 1989; 7: 499-508. |
| [23] | Van Oosterom AT, De Smedt EA, Denis LJ, de Bruijn EA, Mahler C. Suramin for prostatic cancer: a phase I/II study in advanced extensively pretreated disease Eur J Cancer 1990; 26: 422. |
| [24] | La Rocca RV, Stein CA, Danesi R, Myers CE. Suramin, a novel antitumor compound J Steroid Biochem Mol Biol 1990; 37: 893-. |
| [25] | La Rocca RV, Stein CA, Danesi R, Cooper MR, Uhrich M, Myers CE. A pilot study of suramin in the treatment of metastatic renal cell carcinoma Cancer 1991; 67: 1509-3. |
| [26] | Fleck SL, Birdsall B, Babon J, et al. Suramin and suramin analogues inhibit merozoite surface protein-1 secondary processing and erythrocyte invasion by the malaria parasite Plasmodium falciparum J Biol Chem 2003; 278: 47670-77. |
| [27] | Jentsch KD, Hunsmann HH, Nickel P. Inhibition of human immunodeficiency virus type I reverse transcriptase by suramin-related compounds J Gen Virol 1987; 68: 2183-92. |
| [28] | Cadène M, Duranton J, North A, Si-Tahar M, Chignard M, Bieth JG. Inhibition of neutrophil serine proteinases by suramin J Biol Chem 1997; 272: 9950-55. |