- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Phosphorylation State-Dependent High Throughput Screening of the c-Met Kinase
Elham Behshad*, Ronald M. Klabe, Alexander Margulis, Mary Becker-Pasha, Mark J. Rupar, Paul Collier, Phillip C. Liu, Gregory F. Hollis, Timothy C. Burn, Richard Wynn
Abstract
High-throughput screening (HTS) of ~50,000 chemical compounds against phosphorylated and unphosphorylated c-Met, a tyrosine kinase receptor for hepatocyte growth factor (HGF), was carried out in order to compare hit rates, hit potencies and also to explore scaffolds that might serve as potential leads targeting only the unphosphorylated form of the enzyme. The hit rate and potency for the confirmed hit molecules were higher for the unphosphoryalted form of c-Met. While the target of small molecule inhibitor discovery efforts has traditionally been the phosphorylated form, there are now examples of small molecules that target unphosphorylated kinases. Screening for inhibitors of unphosphorylated kinases may represent a complementary approach for prioritizing chemical scaffolds for hit-to-lead follow ups.
Article Information
Identifiers and Pagination:
Year: 2010Volume: 4
First Page: 27
Last Page: 33
Publisher Id: CCGTM-4-27
DOI: 10.2174/1875397301004010027
Article History:
Received Date: 04/2/2010Revision Received Date: 02/3/2010
Acceptance Date: 03/3/2010
Electronic publication date: 23/4/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Incyte Corporation, Applied Technology Group, Experimental Station, Route 141 & Henry Clay Road, Wilmington, DE 19880, USA; Tel: (302) 498-6966; Fax: (302) 425-2721; E-mail: ebehshad@incyte.com
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 04-2-2010 |
Original Manuscript | Phosphorylation State-Dependent High Throughput Screening of the c-Met Kinase | |
INTRODUCTION
There are approximately 500 protein kinases encoded in the human genome. As protein kinases often play critical roles in signaling cascades, they are important regulators of virtually all physiological process. Given that kinase activity is often disregulated in disease processes, protein kinases represent an attractive class of drug targets for small molecule intervention and potentially make up over one quarter of all pharmaceutical drug targets being pursued [1Cohen P. Protein Kinases- the major drug targets of the twenty-first century Nat Rev Drug Discov 2002; 1: 309-15., 2Wesche H, Xiao SH, Young SW. High throughput screening for protein kinase inhibitors Comb Chem High Throughput Screen 2005; 8: 181-95.]. As such, there is much interest in discovering novel small molecule kinase inhibitor scaffolds for pharmaceutical development and drug discovery. Protein kinase domains share a common fold which can be divided into an N-terminal mostly β-strand lobe and a C-terminal mostly α-helical lobe. The so-called activation loop is centrally located between the two kinase lobes. The standard model for kinase activation requires phosphorylation on the activation loop. In general, phosphorylation results in large inter-lobe conformational changes and a significant increase in the catalytic activity of the kinase domain [3Huse M, Kuriyan J. The conformational plasticity of protein kinases Cell 2002; 109: 275-82.]. However, several examples of protein kinases that either do not show activation upon phosphorylation [4Nagar B, Bornmann WG, Pellicena P, et al. Crystal structure of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and Imatinib (STI-571) Cancer Res 2002; 62: 4236-3.] or show significant activity in the unphosphorylated form [5Wang W, Marimuthu A, Tsai J, et al. Structural characterization of autoinhibited c-Met kinase produced by coexpression in bacteria with phosphatase Proc Natl Acad Sci 2006; 103: 3563-68.] are known. While traditional small molecule drug discovery has focused on the phosphorylated “active” form of protein kinases, recently the unphosphorylated “inactive” form is being considered as an attractive form to target for a variety of reasons [6Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations Nat Chem Biol 2006; 2: 358-64.]. For example it is thought that there is considerably more conformational states in unphosphorylated kinases which can then be exploited to achieve selectivity. In particular, it maybe possible to find inhibitors that preferentially bind to conformations only populated by the unphosphorylated form.
c-Met is a tyrosine kinase receptor for hepatocyte growth factor (HGF) that is often deregulated in human cancers making it an attractive drug target. It is a disulfide-linked heterodimeric receptor that results from proteolytic processing of a single chain precursor. Binding of HGF to c-Met results in autophosphorylation of the activation loop at a pair of tandem tyrosine residues (residues 1234 and 1235) and receptor multimerization. This causes increased tyrosine kinase activity, phosphorylation at additional sites in the c-Met cytoplasmic domain of c-Met, recruitment of adaptor proteins and finally downstream activation of various affecters including PI-3-Kinase, PLC-γ, STATs, ERK 1 and 2, and FAK. Activation of c-Met and its downstream pathways result in increased cell growth and motility and decreased apoptosis, which has casually linked this pathway to a wide variety of tumor types [7Corso S, Comoglio PM, Giordano S. Cancer therapy: can the challenge be Met? Trend Mol Med 2005; 11: 284-92.]. As such, c-Met is an attractive candidate for development of anti-tumor therapies [8Liu X, Yao W, Newton RC, Scherle PA. Targeting the c-Met signaling pathway for cancer therapy Expert Opin Investig Drugs 2008; 17: 997-1011.].
Gleevec is a currently approved drug for the treatment of chronic myelogenous leukemia (CML) and is a hallmark targeted therapeutic in oncology [9Fischer OM, Streit S, Hart S, Axel U. Beyond Herceptin and Gleevec Curr Opin Chem Biol 2003; 6: 490-5.]. It has been shown to target the unphosphorylated form of Bcr-Abl [4Nagar B, Bornmann WG, Pellicena P, et al. Crystal structure of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and Imatinib (STI-571) Cancer Res 2002; 62: 4236-3., 10Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of Abelson tyrosine Kinase Science 2000; 289: 1938-42.]. Given the success of Gleevec and the general observation that kinases occupy different conformational states depending upon phosphorylation state, we hypothesized that high throughput screening of the two kinase forms might have different hit rates and identify different lead molecules. We screened a chemical library against phosphorylated and unphosphorylated c-Met to test this hypothesis. The screen yielded a higher hit rate for unphosphorylated c-Met. Several of the hits identified against the unphosphorylated form of c-Met did not measurably inhibit the phosphorylated form. These compounds are potentially good starting points for medicinal chemistry efforts. While future medicinal chemistry will be needed to establish the chemical tractability of these phosphorylation state dependent hits, we discuss other known c-Met inhibitors that appear to be acting on the unphosphorylated form of c-Met in cancer cell lines thus indicating that the approach is feasible in biologically relevant assays. Future screening and chemistry efforts will be needed to see if this approach is more generally applicable to other tyrosine kinases.
MATERIALS AND METHODS
c-Met Cloning, Expression and Purification
Production of phosphorylated and unphosphorylated c-Met: Residue 956 through 1391 (the catalytic domain) of human c-Met was amplified with an N-terminal 6X-HIS tag using the polymerase chain reaction and human liver cDNA. The resulting fragment was cloned into pENTR (Invitrogen, CA), sequence confirmed and transferred to pDEST8 using the Invitrogen Gateway System. Baculovirus was generated with the cMET/pDEST8 clone following the general procedures of the Bac-to-Bac Baculovirus Expression Systems (Invitrogen, CA). E.coli DH10Bac cells were transformed with the cMET/pDEST8 clone and Bacmid DNA was prepared for transfection into Sf21 insect-cells. Conditioned medium containing baculovirus particles was collected from the transfected cells and used for additional infections of Sf21 cells for amplification of the virus. Production of unphosphorylated c-Met was achieved by expressing full length protein tyrosine phosphatase 1B (PTB1B) using an equivalent procedure.
The phosphorylated form of the human c-Met catalytic domain was optimized for expression in Sf21 cells grown in Expression Systems ESF 921 serum-free medium. Cells were infected with baculovirus at a multiplicity of infection (MOI) of 5 and harvested 48 hours post-infection. Unphosphorylated human c-Met (catalytic domain) was produced by baculovirus coinfected with human full-length PTP-1B tyrosine phosphatase at a MOI ratio of 1:1 in Sf21 cells for 48 hours. The degree or lack of phosphorylation was examined by western analysis using both anti-phospho c-Met (pY1234/pY1235, the activation loop phosphorylation sites) and anti-phosphotyrosine antibodies on both the cell lysate and purified protein. The cells were suspended in cold lysis buffer (100 mM potassium phosphate, pH 7.5/250 mM NaCl/5% glycerol/5 mM BME/1% Triton) and lysed by dounce homogenization. The His-tagged c-Met protein was bound to Ni-NTA sepharose, washed with buffer (100 mM potassium phosphate, pH 7.5/250 mM NaCl/5% glycerol/5 mM BME) containing 10 mM and 40 mM imidazole with 100- and 20-column volumes, respectively. His-tagged c-Met was eluted with 10-column volumes of 300 mM imidazole in the same buffer. Eluted protein was dialyzed against the same buffer, concentrated and reloaded on a Talon column. The column was washed with 60- and 10-column volumes of 10 mM and 25 mM imidazole, respectively. c-Met was eluted with 10-column volumes of 150 mM imidazole.
Western Analysis
For the western blotting analysis, unphosphorylated and phosphorylated c-Met samples were resolved on SDS-12% PAGE gel and transferred to PVDF membrane (Invitrogen, CA). Phosphorylation was detected using anti-phosphotyrosine or anti-phospho c-Met (pY1234/pY 1235) (Cell Signal, MA) antibodies using standard protocols.
c-Met Kinase Assay
The assay buffer contained 50 mM Tris-HCl, 10 mM MgCl2, 100 mM NaCl, 0.1 mg/ml BSA, 5mM DTT, pH 7.8. For HTS 0.8 μL of 5 mM of the test compounds dissolved in DMSO were dotted on 384-well plates. DMSO titration suggested that the maximum tolerated concentration of the solvent is 4%. To measure IC50s the compound plate was prepared by 3-fold and 11-point serial dilutions. 0.8 µL of the compound in DMSO was transferred from the compound plate to the assay plate. The final concentration of DMSO was 2%. Solutions of 8 nM unphosphorylated c-Met or 0.5 nM phosphorylated c-Met were prepared in assay buffer. A 1 mM stock solution of peptide substrate Biotin-EQEDEPEGDYFEWLE-amide (Quality Controlled Biochemicals, MA) dissolved in DMSO was diluted to 1 μM in assay buffer containing 400 μM ATP (unphosphorylated c-Met) or 160 uM ATP (phosphorylated c-Met). A 20 μL volume of enzyme solution (or assay buffer for the enzyme blank) was added to the appropriate wells in each plate and then 20 μL/well of substrate solution to initiate the reaction. The plate was protected from light and incubated at 25 °C for 90 min. The reaction was stopped by adding 20 μL of a solution containing 45 mM EDTA, 50 mM Tris-HCl, 50 mM NaCl, 0.4 mg/ml BSA, 200 nM SA-APC and 3 nM EU-Py20. The plate was incubated for 15-30 min at room temperature and HTRF (homogenous time resolved fluorescence) was measured on a Perkin Elmer Fusion α-FP instrument. The HTRF program settings used were as follows: Primary excitation filter 330/30, Primary window: 200 uSec, Primary delay: 50 uSec, Number of flashes: 15, Well read time: 2000 µSec, Secondary excitation filter: 330/30, Secondary window: 400 µSec, Secondary delay: 400 µSec, Primary, Secondary delay: 400 µSec, Primary emission filter: EM 665/10, Secondary emission filter EM 620/10. Percentage of inhibition was calculated for each concentration and IC50 value was generated from curve fitting with GraphPad Prism 3 software. The Km values for ATP were measured in the presence of 1 μ M substrate peptide.
RESULTS AND DISCUSSION
Production and Characterization of c-Met forms
We produced the catalytic domain of c-Met using the baculovirus expression system. Because c-Met has been shown to be a substrate for protein tyrosine phosphatase 1B (PTP1B), unphosphorylated and phosphorylated c-Met was generated in the presence and absence of coexpressed PTP1B, respectively [5Wang W, Marimuthu A, Tsai J, et al. Structural characterization of autoinhibited c-Met kinase produced by coexpression in bacteria with phosphatase Proc Natl Acad Sci 2006; 103: 3563-68.]. Phosphorylation of c-Met was monitored by western analysis using both anti-phospho c-Met (pY1234/pY1235, the activation loop phosphorylation sites) and anti-phosphotyrosine antibodies. Expression of c-Met in the absence of PTP1B produced a kinase that had significant phosphorylation of the activation loop while c-Met expressed in the presence of PTP1B produced no detectable phosphorylation either in the activation loop or elsewhere in the kinase domain (Fig. 1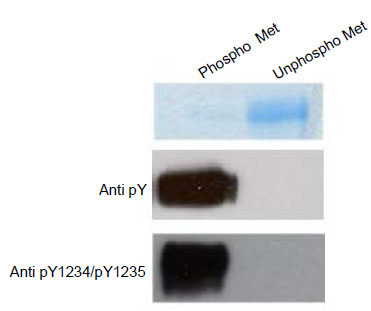 ). Given the 100-fold higher protein load for the unphosphorylated form and lack of detectable phosporylation in that form by western analysis, we conservatively estimate that the level of phosporylation must be less than 0.1%. Similar results have been reported elsewhere using Escherichia coli as an expression host [5Wang W, Marimuthu A, Tsai J, et al. Structural characterization of autoinhibited c-Met kinase produced by coexpression in bacteria with phosphatase Proc Natl Acad Sci 2006; 103: 3563-68.].
). Given the 100-fold higher protein load for the unphosphorylated form and lack of detectable phosporylation in that form by western analysis, we conservatively estimate that the level of phosporylation must be less than 0.1%. Similar results have been reported elsewhere using Escherichia coli as an expression host [5Wang W, Marimuthu A, Tsai J, et al. Structural characterization of autoinhibited c-Met kinase produced by coexpression in bacteria with phosphatase Proc Natl Acad Sci 2006; 103: 3563-68.].

Steady state kinetic analysis was undertaken for each form of the enzyme. The phosphorylated and unphosphorylated forms of c-Met show Km values for ATP of 60 ± 12 and 200 ± 38 μ M, respectively. Relative Kcat values were determined to be 1.0 and 0.065 for phosphorylated and unphosphorylated forms, respectively (Table 1). While traditionally unphosphophoryalted forms of kinases were assumed to be enzymatically inactive several other unphosphorylated kinases have also be reported to be catalytically active [10Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of Abelson tyrosine Kinase Science 2000; 289: 1938-42.-12Giaiwala KS, Wu JC, Christensen J, et al. Kit kinase Mutants show unique mechanisms of drug resistance to Imatinib and Sunitinib in gastrointestinal stromal tumor patients Proc Natl Acade Sci 2009; 106: 1542-47.]. The activity of unphosphorlayted kinases may represent a basal signaling level in cellular environments. The two forms of c-Met are clearly kinetically distinguishable as reflected by effects on both Kcat and Km in steady state kinetic analysis.

While it is theoretically possible that unphosphorylated c-Met undergoes autophosphorylation under the assay conditions, this seems unlikely for several reasons. 1) No phosphorylation was detected by western analysis after incubating this form of the enzyme under assay conditions (data not shown) 2) product formation was linear with time (see Fig. 2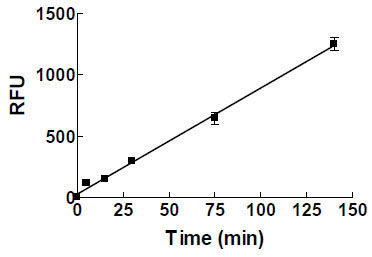 ). Since the phosphorylated enzyme has higher catalytic activity, c-Met phosphorylation during the kinase assay would have resulted in upward curvature for product formation. Timofeevski et al. noted increase in Kcat over time under conditions where autophosphorylation can occur [13Sergei L Timofeevski, Michele A McTigue, Kevin Ryan, et al. Enzymatic characterization of c-Met receptor tyrosine kinase oncogenic mutants and kinetic studies with aminopyridine and triazolopyrazine inhibitors Biochemistry 2009; 48(23): 5339-49.]. No such change is observed under our reaction conditions. 3) As will be shown below, the two forms of the enzyme have reproducible and different IC50 values for a series of inhibitors (Figs. 3A
). Since the phosphorylated enzyme has higher catalytic activity, c-Met phosphorylation during the kinase assay would have resulted in upward curvature for product formation. Timofeevski et al. noted increase in Kcat over time under conditions where autophosphorylation can occur [13Sergei L Timofeevski, Michele A McTigue, Kevin Ryan, et al. Enzymatic characterization of c-Met receptor tyrosine kinase oncogenic mutants and kinetic studies with aminopyridine and triazolopyrazine inhibitors Biochemistry 2009; 48(23): 5339-49.]. No such change is observed under our reaction conditions. 3) As will be shown below, the two forms of the enzyme have reproducible and different IC50 values for a series of inhibitors (Figs. 3A & D). 4) The low concentrations of the kinase in the assay make trans-autophosphorylation unlikely. Timofeevski et al. noted strong c-Met concentration dependence for autophosphorylation [13Sergei L Timofeevski, Michele A McTigue, Kevin Ryan, et al. Enzymatic characterization of c-Met receptor tyrosine kinase oncogenic mutants and kinetic studies with aminopyridine and triazolopyrazine inhibitors Biochemistry 2009; 48(23): 5339-49.] and performed autophosphorylation reactions at 10 uM enzyme (in the presence of 4 mM ATP at 4 °C which can overall lead to ~2-fold decrease in the catalytic activity compared to 200 µM ATP at 25 ºC). Their c-Met concentrations are 2500-fold higher than in the enzyme assays conducted here.
& D). 4) The low concentrations of the kinase in the assay make trans-autophosphorylation unlikely. Timofeevski et al. noted strong c-Met concentration dependence for autophosphorylation [13Sergei L Timofeevski, Michele A McTigue, Kevin Ryan, et al. Enzymatic characterization of c-Met receptor tyrosine kinase oncogenic mutants and kinetic studies with aminopyridine and triazolopyrazine inhibitors Biochemistry 2009; 48(23): 5339-49.] and performed autophosphorylation reactions at 10 uM enzyme (in the presence of 4 mM ATP at 4 °C which can overall lead to ~2-fold decrease in the catalytic activity compared to 200 µM ATP at 25 ºC). Their c-Met concentrations are 2500-fold higher than in the enzyme assays conducted here.

 |
Fig. (3) Comparison of dose-dependent inhibition of phosphorylated and unphosphorylated c-Met by small molecule inhibitors. 4 nM of the unphosphorylated was incubated with 1 µM of peptide substrate, 200 µM ATP and small molecule inhibitors (filled triangles) for 90 minutes at 25 °C. 0.25 nM of the phosphorylated c-Met was mixed with1 µM of peptide substrate 80 µM ATP and small molecule inhibitors (open squares) for 90 minutes at 25 °C. Hit 7 is from Table 2 and Hit 3 is from Table 3. Points are duplicates normalized to 100% for the uninhibited kinase. Data were fit to a hyperbola on logarithmic scale. |
Screening of Small Molecule Libraries
42,000 compounds from the ChemBridge collection were screened against phosphorylated and unphosphorylated c-Met. ChemBridge is a collection of 450,000 verified, druglike, diverse, small molecule compounds. The assay performed well over the course of the screen; control wells for each plate averaged a signal/background ratio of 8.6 with Z´ (Z´ = 1- ([3 X STD negative control] + [3 X STD positive control])/(mean positive control – mean negative control) of 0.74 for the unphosphorylated and 14.5 and 0.81 for the phosphorylated form, respectively. The concentration-response curves for the positive control, SU11274, included on every screening plate, were consistent, with median minimum significant ratio (MSR) value of 1.3 for phosphorylated and 1.2 for unphophorylated [14Brian J, Eastwood BJ, Mark W, et al. The minimum significant ratio: a statistical parameter to characterize the reproducibility of potency estimates from concentration-response assays and estimation by replicate-experiment studies J Biomol Screen 2006; 11: 253-61.]. The Z´ and MSR values for both assays indicate that the screening efforts would be expected to be robust and reliable.
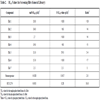
The compounds were tested at 100 µM. All compounds that exhibited greater than 30% inhibition against either form of the enzyme were then retested against both forms by determining their IC50 values. The hits that had been shown to test positive against non-kinase targets from other HTS campaigns were eliminated as false positives (~11% of the total hits). Using a 30 µM cutoff for potency when defining a hit, the hit rate for phosphorylated c-Met was 0.09 % (38 total hits) while the hit rate for unphosphorylated c-Met is 0.13% (56 total hits). The hit rate for unphosphorylated c-Met was ~ 44% higher than for phosphorylated c-Met. While this ratio of hit rates might change if larger numbers of compounds were screened, it is clear that for the limited set tested there is a higher number of potential lead molecules generated when screening with the unphosphorylated form of the enzyme. Inspection of the relative potencies for compounds indicates that the majority of compounds are roughly equally potent on both forms of c-Met with ratios close to 1.0 (Fig. 3C). There are however, compounds that exhibit at least a 5-fold more potent IC50 value for unphosphorylated c-Met relative to phosphorylated c-Met (Table 2). In fact of the 56 hits for unphosphorylated c-Met, six have greater than 5-fold specificity. These hits (ratios >5), four of which with purine moities and two of which with a novel benzyloxybenzaldehyde oxime core structure, represent potential lead molecules that would not have been identified by screening only the phosphorylated form of c-Met. On the other hand, no compound shows significantly greater potency on the phosphorylated form (ratio significantly <1.0) which accounts for the increased hit rate for the unphosphorylated form. From a structural perspective, the specific molecules must be binding to a conformation that only the unphosphorylated form can easily adopt. The nonspecific molecules, on the other hand, must bind to a conformation that is present in both forms of the enzyme. The scenario is quite plausible given the known large conformation changes that kinases undergo and that the conformation preferences are in large part determined by phosphorylation state [15Huse M, Kuriyan J. The conformational plasiticity of protein kinases Cell 2002; 107: 275-.].
We also examined two published kinase inhibitors that inhibit c-Met. Staurosporine is a well known ATP-competitive nonspecific kinase inhibitor. SU11274 is a commercially available potent c-Met inhibitor. SU11274 is a well studied specific ATP-competitive inhibitor and has been shown to be active against numerous cancer cell lines [16Sattler M, Pride YB, Ma P, et al. A novel small molecule met inhibitor induces apoptosis in cells transformed by the oncogenic TPR-MET tyrosine kinase Cancer Res 2003; 63(17): 5462-9.-18Bellon SF, Kaplan-Lefko P, Yang Y, et al. Dussault. c-Met inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma-related mutations J Biol Chem 2008; 283(5): 2675-83.]. Table 2 and Figs. (3A and 3B) show the IC50 values for these two inhibitors. Staurosporine has essentially the same IC50 value for both forms of the enzyme while SU11274 is 5 nM on the unphosphorylated form and 275 nM on the phosphorylated form, a 55-fold difference in potency. Furthermore, the IC50 for SU11274 is 30 nM in SNU5 proliferation assays. SNU5 is a gastric cancer cell line in which c-Met is amplified. Given that the compound is more potent on cells than on the phosphorylated form of the enzyme, it is likely that the cellular activity is being driven by inhibition of the unphosphorylated enzyme. The discrepancy is even larger when one accounts for the high cellular ATP concentrations of approximately 1 mM (predicted IC50 values for SU11274 are 30 nM and 3331 nM for the phosphorylated and unphosphorylated forms, respectively). We have noted the same trend with a large number of compounds in our internal c-Met program.
The observations made during our screening endeavor and examination of known c-Met inhibitors convinced us to examine our internal kinase library for additional inhibitors of the unphosphorylated form. The internal library is a compound collection that resulted from medicinal chemistry efforts on several unrelated kinase targets. All chemical series were shown to be ATP competitive against their respective kinase target.
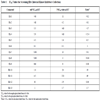
Using a 30 µM cutoff for potency when defining a hit, the hit rate for phosphorylated c-Met was 1.0 % (73 total hits) while the hit rate for unphosphorylated c-Met was 0.89 % (62 total hits). The hit rate for unphosphorylated c-Met was similar to the phosphorylated c-Met. While screening a random library led us discover potential lead molecules that inhibit unphosphorylated c-Met, screening our internal kinase inhibitors (Table 3) resulted in identifying seven compounds that were 5-fold or more potent against the phosphorylated form (ratio significantly <1.0). For example, while Hit 1 and 2 inhibit the phosphorylated form with IC50s of 11 and 4.2 µM respectively, they do not inhibit the unphosphorylated form. There are also compounds which do not inhibit the phosphorylated form but are relatively potent against unphosphorylated c-Met (Hit 3 and 4). In fact six compounds from the 62 hits were identified to be 5-fold or more selective for unphosphorylated form. The screening of our internal kinase collection has shown that it is possible to obtain compounds that are more potent on the phosphorylated c-Met. Most of these compounds had a common core of benzoimidazo (or thiozolo) isoquiolinon, a known kinse scaffold. These compounds presumably bind a conformation (or induce a conformation) that is preferred by the phosphorylated form and/or disfavored by the unphosphorylated form.
CONCLUSION
We have shown that screening the unphosphorylated and phosphorylated form of c-Met yields different hit molecules. Screening of random and kinase libraries, it was possible to find molecules that are equipotent on both forms, more potent on the unphosphorylated form and more potent on the phosphorylated form. The specific inhibitors likely exploit different conformational preferences between the two forms of c-Met. As is well known, kinases undergo large conformational changes upon phosphorylation [15Huse M, Kuriyan J. The conformational plasiticity of protein kinases Cell 2002; 107: 275-.]. Structural studies would be needed to examine this further in the case of the inhibitors explored here. However, there is precedent for this approach. Gleevec is an Abl kinase inhibitor that is approved for the treatment of chronic myelogenous leukemia (CML) [19Nadal E, Olavarria E. Imatinib mesylate (Gleevec/Glivec) a molecular-targeted therapy for chronic myeloid leukaemia and other malignancies Int J Clin Pract 2004; 58(5): 511-6.]. It has been shown that Gleevec is significantly more potent on the unphosphorylated from of Abl kinase and that the inhibitor binds to an inactive conformation that only unphosphorylated Abl can easily adopt [4Nagar B, Bornmann WG, Pellicena P, et al. Crystal structure of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and Imatinib (STI-571) Cancer Res 2002; 62: 4236-3., 10Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of Abelson tyrosine Kinase Science 2000; 289: 1938-42.]. Interestingly, Abl has also been shown to have activity in the unphosphorylated form [4Nagar B, Bornmann WG, Pellicena P, et al. Crystal structure of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and Imatinib (STI-571) Cancer Res 2002; 62: 4236-3., 10Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of Abelson tyrosine Kinase Science 2000; 289: 1938-42.]. Gleevec also falls into “type II” class of inhibitors [6Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations Nat Chem Biol 2006; 2: 358-64.]. This class of inhibitors contains a hydrogen bond donor-acceptor pair (amide or urea) and a hydrophobic tail that interact with the “allosteric site”. Most of them also contain a “head“ group that extends into the adenine region. They have been shown to bind to the unphosphorylated form and induce or stabilize an inactive conformation. While Gleevec falls into this category there are examples of type I inhibitors that bind to the unphosphorylated form more tightly. SU11274 is one example which was discussed earlier and AM7 is another. AM7, (5-(3-fluoro-4-((6-(methyloxy)-7-((3-(4 morpholinyl)prpyl)oxy)-4-quinolinyl) oxy)phenyl)-3-methyl-2-(phenylmethyl)-4(3H)-pyrimidin-one) developed by Amgen, was shown to be more than 3-fold more potent on unphosphorylated Met (IC50 of 5 vs. 17 nM)18. Interestingly the binding mode of AM7 seems to be different from SU11274 (which is also different from how a type I inhibitor would bind). They both bind to the kinase linker but AM7 extends into the C-helix hydrophobic region which is 8 Å away from the DFG pocket. While SU11274 and AM7 bind to the unphosphorylated form more tightly, Pan and co-workers recently identified MK2461 that binds to the phosphorylated enzyme 6-fold tighter [20Pan B, Chan KY, Chenard M, et al. MK-2461, a Novel multitargeted kinase inhibitor, preferentially inhibits the activated c-Met receptor Cancer Res 2010; 70(4): 1524-33.]. These examples illustrate that there exists many conformations available to both phosphorylated and unphosphorylated kinases.
The results presented above show that different hit molecules are obtained by screening the phosphorylated and unphosphorylated forms of c-Met kinase. Future research will be required to determine the tractability of these particular hits. While it is unlikely that all of these hits can be sufficiently pursued, there is clearly a number of potential new scaffolds when all screening hits are evaluated. It is intriguing to speculate that this approach could be employed to target other kinases; however, this methodology is perhaps more productive for kinases that have some characteristics similar to c-Met. As noted above, c-Met has significant catalytic activity in the unphosphorylated form. This may indicate that structural characteristics that allow for inhibition in the unphosphorylated form may not be present in kinases without activity prior to phosphorylation. It is notable that Gleevec, a marketed drug for AML, is the prototypical inhibitor of an unphosphorylated kinase target and that its target, Abl, has catalytic activity that is little changed upon phosphorylation of its activation loop [10Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of Abelson tyrosine Kinase Science 2000; 289: 1938-42.]. Additionally, it may be easier to translate activity against an unphosphorylated form from an enzyme assay to a cellular context by targeting kinases that autophosphorylate, typically tyrosine kinases. In this context, during the continual autophosphorylation and dephosphorylation by protein phosphatases, binding to the unphosphorylated form of a kinase may shift the “equilibrium” to the unphosphorylated, generally less active, form. Should the activity present in unphosphorylated kinases translate to a basal level of signaling in cellular contexts, binding of the unphosphorylated form may also reap additional pharmacological benefit as inhibitors that don’t bind this form would not impact this basal level of signaling. This scenario is unlikely to apply for kinases that are part of kinase cascades and are phosphorylated by upstream, often unrelated, kinases. These are more typically serine/threonine kinases. Future studies will be required to address these issues. It should also be pointed out that it may be possible to initiate chemistry efforts on compounds that are specific for an unphosphorylated form but build in potency for the phosphorylated form during the medicinal chemistry optimization process. Screening the unphosphorylated form of a kinase can provide a richer ensemble of screening hits for this type of approach.