- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
High-Throughput Multiplexed Quantitation of Protein Aggregation and Cytotoxicity in a Huntington’s Disease Model
Steven A Titus, Noel Southall, Juan Marugan, Christopher P Austin , Wei Zheng*
Abstract
A hallmark of Huntington’s disease is the presence of a large polyglutamine expansion in the first exon of the Huntingtin protein and the propensity of protein aggregation by the mutant proteins. Aberrant protein aggregation also occurs in other polyglutamine expansion disorders, as well as in other neurodegenerative diseases including Parkinson’s, Alzheimer’s, and prion diseases. However, the pathophysiological role of these aggregates in the cell death that characterizes the diseases remains unclear. Identification of small molecule probes that modulate protein aggregation and cytotoxicity caused by aggregated proteins may greatly facilitate the studies on pathogenesis of these diseases and potentially lead to development of new therapies. Based on a detergent insoluble property of the Huntingtin protein aggregates, we have developed a homogenous assay to rapidly quantitate the levels of protein aggregates in a cellular model of Huntington’s disease. The protein aggregation assay has also been multiplexed with a protease release assay for the measurement of cytotoxicity resulting from aggregated proteins in the same cells. Through a testing screen of a compound library, we have demonstrated that this multiplexed cytotoxicity and protein aggregation assay has ability to identify active compounds that prevent cell death and/or modulate protein aggregation in cells of the Huntington’s disease model. Therefore, this multiplexed screening approach is also useful for development of high-throughput screening assays for other neurodegenerative diseases involving protein aggregation.
Article Information
Identifiers and Pagination:
Year: 2012Volume: 6
First Page: 79
Last Page: 86
Publisher Id: CCGTM-6-79
DOI: 10.2174/1875397301206010079
Article History:
Received Date: 8/10/2012Revision Received Date: 5/11/2012
Acceptance Date: 5/11/2012
Electronic publication date: 28/12/2012
Collection year: 2012
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the National Center for Advancing Translational Sciences/NIH, 9800 Medical Center Drive, MSC: 3370, Bethesda, MD 20892-3370, USA; Tel: (301)827-6727; Fax: (301)217-5728; E-mail: wzheng@mail.nih.gov
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 8-10-2012 |
Original Manuscript | High-Throughput Multiplexed Quantitation of Protein Aggregation and Cytotoxicity in a Huntington’s Disease Model | |
INTRODUCTION
Protein aggregation occurs in several neurodegenerative disorders including Huntington’s disease, Alzheimer's disease, Parkinson's disease, Amyotrophic Lateral Sclerosis, and Prion disease. Huntington’s disease has been reported to be caused by an abnormally long expansion of a CAG trinucleotide repeat located in exon 1 of the Huntington (HTT) gene although many other factors have been investigated [1 Varma H. Drug screening for Huntington's disease and other neurodegenerative disorders Curr Mol Pharmacol 2010; 3(3 ): 164-73.]. The expansion of CAG trinucleotide results in a HTT protein bearing a long stretch of polyglutamine residues (PolyQ). The severity and time of onset of the disease are directly proportional to length of the PolyQ expansion [2 Walker FO. Huntington's disease Lancet 2007; 369(9557 ): 218-8.]. The formation of intranuclear inclusions by the HTT protein with long PolyQ expansion is a characteristic hallmark of the disease. While the question of whether the aggregates in cells are toxic or even protective is still debated [3 Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death Nature 2004; 431(7010 ): 805-10., 4 Imarisio S, Carmichael J, Korolchuk V, et al. Huntington's disease: from pathology and genetics to potential therapies Biochem J 2008; 412(2 ): 191-209.], small molecule modulators that prevent the formation of protein aggregates should be useful research tools for further exploring the pathology of the disease as well as serving as lead compounds for potential drug development.
Existing assays for measuring protein aggregation are limited to lower-throughput or non-quantitative methods [5 Apostol BL, Kazantsev A, Raffioni S, et al. A cell-based assay for aggregation inhibitors as therapeutics of polyglutamine-repeat disease and validation in Drosophila Proc Natl Acad Sci USA 2003; 100(10 ): 5950-.-8 Pollitt SK, Pallos J, Shao J, et al. A rapid cellular FRET assay of polyglutamine aggregation identifies a novel inhibitor Neuron 2003; 40(4 ): 685-94.]. Protein aggregates in cells are commonly detected by a direct staining with a fluorescent dye such as Congo Red [9 Scherzinger E, Lurz R, Turmaine M, et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo Cell 1997; 90(3 ): 549-8.], immunostaining with an antibody against to the aggregated protein [10 Gutekunst CA, Li SH, Yi H, et al. Nuclear and neuropil aggregates in Huntington's disease: Relationship to neuropathology J Neurosci 1999; 19(7 ): 2522-34.], expression of an epitope tag, or a fluorescent protein-tagged fusion protein. Many protein aggregation assays require microscopic or an imaging-based instrument for detection and thus the throughput for compound screening is relatively low. Although imaging instrumentation for high content screening (HCS) has advanced remarkably in the last decade, the limited speed of data acquisition and data processing prevent HCS from being used in protein aggregation assays for screening of very large compound collections [11 Scotter EL, Narayan P, Glass M, Dragunow M. High throughput quantification of mutant huntingtin aggregates J Neurosci Methods 2008; 171(1 ): 174-9., 12 Varma H, Lo DC, Stockwell BR. High throughput screening for neurodegeneration and complex disease phenotypes Comb Chem High Throughput Screen 2008; 11(3 ): 238-48.]. Here we report a novel homogenous protein aggregation assay using a laser scanning cytometer plate reader to quantitate the protein aggregates formed in the cells by expression of GFP-PolyQ fusion proteins. This assay has been multiplexed with a cytotoxicity assay for sequential measurements of cell viability and protein aggregates in a cell model of Huntington’s disease that has been used in a testing screen of a compound library. Our results indicate that this multiplexed assay for protein aggregation and cytotoxicity is a robust and reliable assay method for high throughput screening.
MATERIALS AND METHODS
Materials
Tebufenozide and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). DMEM medium and other cell culture supplies were obtained from Invitrogen (Carlsbad, CA). The 1536-well compound plates and black/clear assay plates were purchased from Greiner Bio-one (Monroe, NC). The CytotoxGlo protease release assay was purchased from Promega (Madison, WI), and the ATP-Lite assay was purchased from PerkinElmer (Waltham, MA).
Cell Culture and Plating
Rat pheochromocytoma PC12 cell lines harboring HTT-Q103 or Q25 (a wild type control) fused to GFP under a tebufenozide inducible promoter were kindly provided by Dr. Eric Schweitzer [12 Varma H, Lo DC, Stockwell BR. High throughput screening for neurodegeneration and complex disease phenotypes Comb Chem High Throughput Screen 2008; 11(3 ): 238-48., 13 Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington's disease Neurobiol Dis 2004; 16(3 ): 546-5.]. Cells were maintained in the phenol red free DMEM medium at 37γC under a humidified atmosphere containing 5% CO2 and 95% air. The medium contained 5% calf serum, 5% horse serum (both from HyClone, Logan UT), 250 µg/ml geneticin, 1 ( pen/strep, and 2 mM L-glutamine. Cells were passaged when they reached 80 to 90 % confluency. The PC-12 cell lines have stringent growth conditions; if the cells become too confluent over time, the entire population will perish, and if the cells are seeded too sparsely, they will not grow efficiently. Phenol red was shown to slightly inhibit the luminescent signals so phenol red free media was chosen for the assay.
Cells were plated at a density of 1000 to 1500 cells/well in black, clear bottom, tissue culture treated and low base, 1536 well plates in 5 µl/well of complete DMEM media except geneticin using a Multidrop Combi dispenser (Thermo Scientific, Waltham, MA) and incubated for 24 hr at 37(C under a humidified atmosphere containing 5% CO2. Tebufenozide was used at 200 nM final concentration to induce expression of Q103-GFP and Q25-GFP fusion proteins in these PC12 cell lines. Both tebufenozide and test compounds in DMSO solution were sequentially added to the assay plates at a volume of 23 nl/well using a Kalypsys pintool station. The final concentration of DMSO in assay plates was 0.46% and no significant cytotoxicity was observed in cells treated with this concentration of DMSO (data not shown). The assay plates were incubated at 37(C for 48 hours before being assayed in the following multiplexed dual read assay format.
Measurement of Cytotoxicity Caused by Protein Aggregation
Significant cytotoxicity and subsequent aggregation of the Q103-GFP fusion proteins were found after the induction of the fusion protein expression in the PC12 cell line. Cytotoxicity was first determined by a CytotoxGlo protease release assay kit by quantitating the activity of proteases released from impaired cell membrane. Briefly, 2ul/well of the reagent mixture from the protease assay kit was added to assay plates and the plates were centrifuged at 1500 RPM for 30 sec to remove air bubbles which would affect measurement of the luminescent signal. After 10 minutes of incubation at room temperature, the assay plates were measured in a luminescent mode of Viewlux plate reader for the activity of protease released to the media. The protease activity in the media increases with the increased damaged integrity of plasma membrane due to the cytotoxicity of protein aggregation in the Q103-GFP PC12 cell line.
Measurement of Protein Aggregates
Cells in the same assay plates after the above cytotoxicity measurement were then treated with 1 µl/well of Triton-X100 (0.25% final) for cell lysis that dissolved soluble proteins from the bottom of assay plates. Only the aggregated proteins that were insoluble to the detergent treatment still attached to the bottom of assay plates after this detergent treatment. The assay plates were then measured by a bottom reading mode in an Acumen Explorer reader (TTP Labtech Inc., Melbourn, UK), a laser scanning cytometer, for quantitation of the numbers of Q103-GFP aggregates. The measurement parameters in the Acumen Explorer reader included (1) excitation with a 488 nm laser and emission at 500-530 nm, (2) PMT voltage of 450 volts, and (3) the number of fluorescent objects in size of 1 to 25 µm with a signal of 6 standard deviations above background. The number of fluorescent objects was directly proportional to the severity of protein aggregation observed under a fluorescence microscope in the Q103-GFP cell line.
Compound Libraries
A library collection of 220,581 compounds was screened at four compound concentrations. All compounds were dissolved in DMSO at 10 mM as stock solutions that were serially diluted in DMSO in 384-well plates at a ratio of 1:5. The four sets of 384-well inter-plate dilution plates were subsequently reformatted into one set of 1536-well compound plates at 7 µl/well using a Cybi-well dispensing station with a 384-well head (Cybio, Inc.). The final compound concentrations in 5 µl/well assay volume ranged from 500 nM to 40 µM in our screen.
Instruments for Liquid Handling and Plate Detection
The reagent dispensing and compound transfer in 1536-well plates were handled by automated work stations. A BioRAPTR work station (Beckman Coulter, Brea, CA) using solenoid valves for dispensing was employed to deliver reagents to 1536-well plates ranging from 0.5 to 5 µl per well. A pintool compound transfer station was used to transfer 23 nl compounds in DMSO solution to the media of 5 µl/well cells in 1536-well assay plates.
Data Analysis
The primary screening data was first analyzed using customized software developed internally [21 Wang Y, Jadhav A, Southal N, Huang R, Nguyen DT. A grid algorithm for high throughput fitting of dose-response curve data Curr Chem Genom 2010; 4: 57-66.]. Clustering analysis of active compounds from the primary screening was performed with Leadscope® Hosted Client (Leadscope Inc., Columbus, OH). The results of concentration response curves were analyzed and plotted with the Prism program (GraphPad Software, San Diego, CA).
RESULTS
Measurement of Protein Aggregates
The Q103-GFP cell line has been used in previous screening assays as a cellular model of HD [12 Varma H, Lo DC, Stockwell BR. High throughput screening for
neurodegeneration and complex disease phenotypes Comb Chem
High Throughput Screen 2008; 11(3
): 238-48., 13 Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs
to treat Huntington's disease Neurobiol Dis 2004; 16(3
): 546-5.]. We used a PC12 cell line that inducibly expresses the first 17 amino acids of exon 1 of the HTT gene with 103 glutamines fused to a GFP reporter. The induction of Q103-GFP expression in the cells causes the formation of perinuclear, fluorescent aggregates (Fig. 1A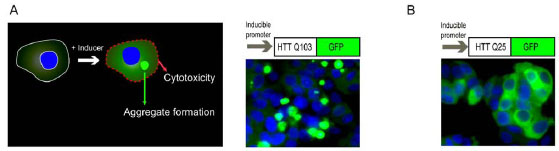 ). The size and fluorescence intensity of these aggregates increase with cell incubation time in the presence of inducer. Additionally, expression of the Q103-GFP fusion protein is cytotoxic, resulting in approximately 40-50% cell death after 48 hr induction of expression of the fusion protein [13 Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs
to treat Huntington's disease Neurobiol Dis 2004; 16(3
): 546-5.]. However, the protein aggregation and resultant cytotoxicity are not observed in the cells of a control line with Q25-GFP fusion protein (Fig. 1B) after 48 hr incubation with the inducer.
). The size and fluorescence intensity of these aggregates increase with cell incubation time in the presence of inducer. Additionally, expression of the Q103-GFP fusion protein is cytotoxic, resulting in approximately 40-50% cell death after 48 hr induction of expression of the fusion protein [13 Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs
to treat Huntington's disease Neurobiol Dis 2004; 16(3
): 546-5.]. However, the protein aggregation and resultant cytotoxicity are not observed in the cells of a control line with Q25-GFP fusion protein (Fig. 1B) after 48 hr incubation with the inducer.

It is challenging to measure these protein aggregates in a high throughput screening format as all existing assays require extensive manipulation of the samples including several complicated assay steps. During the course of assay development, we observed that the fluorescent GFP aggregates in Q103-GFP cells remained on the bottom of assay plates after cell lysis by 0.25% Triton X-100. In contrast, no fluorescent signal was observed on the bottom of assay plates with the Q25-GFP control cells after the same detergent treatment (Fig. 2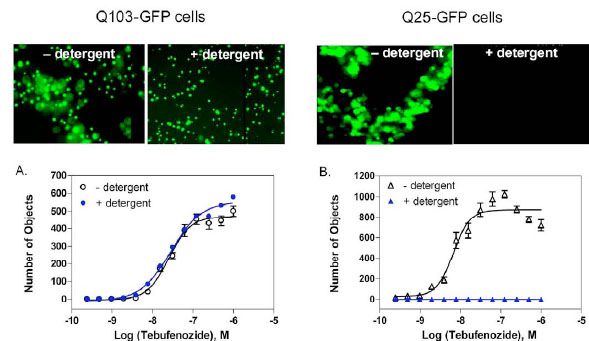 ). The results indicate that the soluble fusion proteins were released into the solution after the detergent treatment, while the protein aggregates were insoluble to detergent and remained on the bottom of assay plates. Importantly, this finding enabled us to develop a homogenous assay for the rapid quantitation of protein aggregates without any cell wash steps. In addition, we found that our observation is similar to a previous report that the HTT polyQ aggregates were resistant to detergent treatment whereas non-aggregated polyQ proteins were solublized by detergent [14 Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman
D. Insoluble detergent-resistant aggregates form between
pathological and nonpathological lengths of polyglutamine in
mammalian cells Proc Natl Acad Sci USA 1999; 96(20
): 11404-9.].
). The results indicate that the soluble fusion proteins were released into the solution after the detergent treatment, while the protein aggregates were insoluble to detergent and remained on the bottom of assay plates. Importantly, this finding enabled us to develop a homogenous assay for the rapid quantitation of protein aggregates without any cell wash steps. In addition, we found that our observation is similar to a previous report that the HTT polyQ aggregates were resistant to detergent treatment whereas non-aggregated polyQ proteins were solublized by detergent [14 Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman
D. Insoluble detergent-resistant aggregates form between
pathological and nonpathological lengths of polyglutamine in
mammalian cells Proc Natl Acad Sci USA 1999; 96(20
): 11404-9.].

We also found that the fluorescence intensities from whole wells of assay plates measured by a conventional plate reader were not able to detect the difference between the Q103-GFP and Q25-GFP control cells. This could be due to the similar expression levels of total GFP fusion proteins in both cell lines and the fluorescence intensities from soluble proteins and aggregates were not distinguishable by measuring a whole well in assay plates. We then used a laser scanning plate cytometer (Acumen Explorer reader) which read from the bottom of assay plates and applied a signal threshold algorithm to identity fluorescent objects (e.g. aggregates) on the bottom of plates against the fluorescent background in solution. We found that the GFP tagged aggregates in Q103-GFP cells were distinguishable from the soluble GFP fusion proteins in Q25-GFP control cells after the detergent treatment. Thus, from optimizing the measurement parameters, only the intensity of fluorescent objects ranging in size of 1 to 25 µm were counted from the bottom of assay plates. The EC50 values of tebufenozide, an inducer for the fusion protein expression, were 25.5 and 28.9 nM in the Q103-GFP cells prior to and after the detergent treatment, respectively (Fig. 2A). The result indicated that the detergent treatment did not change the fluorescence signals from Q103-GFP fusion proteins. Although the concentration response of tebufenozide in Q25-GFP cells was observed before the detergent treatment, no detectable signal was found by using the Acumen Explorer reader because the Q25-GFP fusion proteins were dispersed in solution after the detergent treatment (Fig. 2B). Thus, this protein aggregation assay based on the laser scanning cytometer detection is able to distinguish the aggregated fusion proteins from the un-aggregated proteins in assay plates after the treatment with 0.25% Triton X-100 detergent.
Measurement of Cytotoxicity with a Protease Release Assay
Expression of long polyglutamine expansions such as in the Q103-GFP cells has been shown to be a cause of protein aggregation and resulting cell death in many cell types [15 Li SH, Li XJ. Huntingtin and its role in neuronal degeneration Neuroscientist 2004; 10(5 ): 467-75.]. Compounds that inhibit the cytotoxicity of polyglutamine expression are of interest as research tools to study the pathophysiology of many diseases related to protein aggregation. In order to identify lead compounds which inhibit cytotoxicity caused by expression of long polyglutamine expansions, we multiplexed the assay readout to measure both protein aggregation and cytotoxicity in the same well of the same assay plate.
In Q103-GFP cells, only a subpopulation of cells (usually ~40-50%) is adversely affected after 48 hr induction of expression of Q103-GFP proteins. Previous reports indicated that signal-to-basal ratios of MTT or LDH release assays in these cells were not robust with lower signal-to-basal ratios [13 Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs
to treat Huntington's disease Neurobiol Dis 2004; 16(3
): 546-5.]. We initially tried an ATP content assay for the measurement of ATP levels in viable cells with a luminescence readout. The signal-to-basal ratio was around 1.5 to 2 fold, presumably due to the large percentage of viable cells (Fig. 3A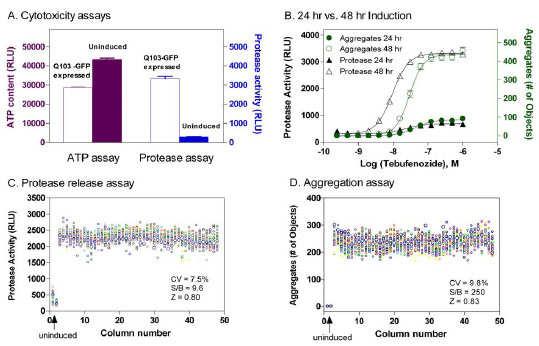 ). We then applied a protease release assay that measures the activity of released proteases from damaged plasma membrane due the toxicity of Q103-GFP proteins. We found that the signal-to-basal ratio of this protease release assay in Q103-GFP cells was 11.1 fold, indicative of a robust assay for high throughput screening (Fig. 3A). The high signal-to-basal ratio was found in this protease release assay that may be due to the lower basal signal in this assay compared with the high basal signal in the ATP content assay [16 Cho MH, Niles A, Huang R, et al. A bioluminescent cytotoxicity
assay for assessment of membrane integrity using a proteolytic
biomarker Toxicol in Vitro 2008; 22(4
): 1099-6.]. The cellular proteases only leak out to media when the integrity of plasma membrane is perturbed in apoptotic or necrotic cells. The cytotoxicity of GFP-Q103 protein expression after 48 hr induction with the inducer tebufenozide was much more significant compared with that of 24 hr induction, which correlated with the time course of protein aggregation formation detected by the above assay (Fig. 3B).
). We then applied a protease release assay that measures the activity of released proteases from damaged plasma membrane due the toxicity of Q103-GFP proteins. We found that the signal-to-basal ratio of this protease release assay in Q103-GFP cells was 11.1 fold, indicative of a robust assay for high throughput screening (Fig. 3A). The high signal-to-basal ratio was found in this protease release assay that may be due to the lower basal signal in this assay compared with the high basal signal in the ATP content assay [16 Cho MH, Niles A, Huang R, et al. A bioluminescent cytotoxicity
assay for assessment of membrane integrity using a proteolytic
biomarker Toxicol in Vitro 2008; 22(4
): 1099-6.]. The cellular proteases only leak out to media when the integrity of plasma membrane is perturbed in apoptotic or necrotic cells. The cytotoxicity of GFP-Q103 protein expression after 48 hr induction with the inducer tebufenozide was much more significant compared with that of 24 hr induction, which correlated with the time course of protein aggregation formation detected by the above assay (Fig. 3B).

Multiplexing Cytotoxicity Measurement with Detection of Protein Aggregation
Because the measurements of cytotoxicity and protein aggregation described in the above experiments utilize two different detection modes (e.g. luminescence and fluorescence intensity, respectively), we tried to multiplex them into sequential measurements in a single assay plate. The main advantage for using the same assay plate to measure compound effects on both cytotoxicity and protein aggregation is that the compound activities in these two assays are determined from the cells treated under identical experimental conditions. The results from such a multiplexed assay would be more comparable for the similarity or difference in compound activities on protein aggregation and cytotoxicity caused by the aggregated proteins. This multiplexed assay also has very few steps and is therefore relatively simple for the robotic screening (Table 1). Briefly, the cells are seeded into a 1536 well plate and incubated for one day. Expression of the Q103-GFP or Q25-GFP fusion proteins is induced by the addition of 200 nM tebufenozide (with or without library compound addition). After a 48 hr treatment with the inducer, cytotoxicity is first measured by the protease release assay in the luminescence detection mode with a single reagent addition step. After the cytotoxicity measurement, the assay plates are treated with detergent to lyse the cells that subsequently allows all soluble proteins to diffuse into the surrounding media. The fluorescent protein aggregates are then measured using a laser scanning plate cytometer (Fig. 4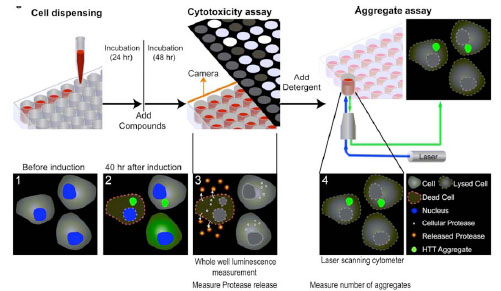 ).
).

Assay Validation and Compound Library Screen
Using a DMSO plate as a control , we found that the signal-to-basal ratios for cytotoxicity and protein aggregate measurements were 9.6 and 250 fold, the CVs were 7.5% and 9.8%, and the Z’ factors were 0.80 and 0.83, respectively (Figs. 3C and 3D). The results from this multiplexed assay demonstrate the robustness of assay performance, which is suitable for high throughput screening of large compound collections. We then screened a library of 220,581 small molecule compounds at four concentrations ranging from 0.5 to 40 µM. Several groups of active compounds with different effects on cytotoxicity and protein aggregation were identified from this screen. The primary screening results have been deposited in PubChem (AID 1471, 1482 and 1688).
Representative compounds identified from the compound library screening are shown in Fig. (5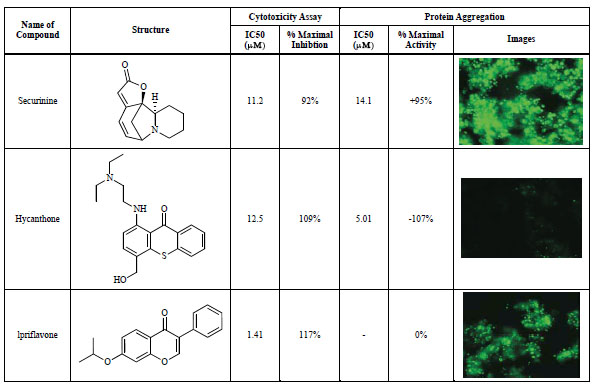 ). The cytoprotective compounds identified were classified into three groups; (1) compounds inhibiting cell death but increasing protein aggregation (top panel), (2) compounds inhibiting both cell death and protein aggregation (middle panel), and (3) compounds inhibiting cell death without an effect on protein aggregation (bottom panel). While all three representative compounds exhibited cytoprotective effect in the protease release assay as well as an ATP quantitation assay (data not shown), compound 1 (securinine) increased the number of protein aggregates (green color in the image), compound 2 (hycanthone) decreased the number of protein aggregates and compound 3 (ipriflavone) had no effect on the protein aggregates compared with the control cells. These active compounds could be useful research tools for the further studies on the mechanism of protein aggregation and its relation with cytotoxicity as well as further studies in animal models. Some of these compounds are currently under investigation to identify their potential mechanisms of action. The results of structure-activity studies as well as the potential mechanism of action will be reported in a separate article.
). The cytoprotective compounds identified were classified into three groups; (1) compounds inhibiting cell death but increasing protein aggregation (top panel), (2) compounds inhibiting both cell death and protein aggregation (middle panel), and (3) compounds inhibiting cell death without an effect on protein aggregation (bottom panel). While all three representative compounds exhibited cytoprotective effect in the protease release assay as well as an ATP quantitation assay (data not shown), compound 1 (securinine) increased the number of protein aggregates (green color in the image), compound 2 (hycanthone) decreased the number of protein aggregates and compound 3 (ipriflavone) had no effect on the protein aggregates compared with the control cells. These active compounds could be useful research tools for the further studies on the mechanism of protein aggregation and its relation with cytotoxicity as well as further studies in animal models. Some of these compounds are currently under investigation to identify their potential mechanisms of action. The results of structure-activity studies as well as the potential mechanism of action will be reported in a separate article.

DISCUSSION
Our goal was to develop an HTS assay that has an ability to interrogate compound effects on both the poly-Q aggregation and resulting cytotoxicity in a cell model of HD. The protein aggregation assay described here has several advantages over previous assays using a standard or confocal fluorescence microscope. First, the high energy excitation used in the laser scanning cytometer significantly improves the signal-to-basal ratio of the measurement. Second, the read time of ~10 minutes per plate is significantly shorter compared to an automated imaging system which typically requires 45 or more minutes to measure a 1536 well plate. Third, the data processing is conducted in parallel with the image acquisition so no extra time was required for data processing. Fourth, the size of data file for an assay plate, typically 500 kb, is much smaller compared to the image file generated by a microscopic reader. In contrast, the imaging-based HCS screens capture multiple image fields per well which in turn produces large data files; typically 4 to 10 gigabytes per 1536 well plate. Therefore, this protein aggregation assay is convenient and user-friendly for compound screening of large libraries.
The phenotype of toxic gain of function associated with polyglutamine expansions is similarly present in all CAG expansion-related diseases [17 Orr HT, Zoghbi HY. Trinucleotide repeat disorders Ann Rev Neurosci 2007; 30: 575-621.]. The formation of protein aggregates observed in these diseases is a slow process whereby the protein filaments slowly undergo conformational changes; presumably evolving from compact b-sheets, to protofibrils, fibrils, and then aggregate foci, and finally visible aggregates [18 Bates G. Huntingtin aggregation and toxicity in Huntington's disease Lancet 2003; 361(9369): 1642-4.]. These stages appear to have differential affects on cytotoxicity [19 Bucciantini M, Giannoni E, Chiti F, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases Nature 2002; 416(6880): 507-11.]. However, it is still unknown at which stage these proteins become detergent insensitive, and how that relates to cytotoxicity. Future studies exploring various parameters such as different time points, detergent concentrations, or different types of detergents may help to better understand of the process and pathogenesis of aggregation formation.
Although 220,581 compounds were screened using this multiplex assay and several compounds identified, an extended library screen with a larger or more diverse compound collection might be needed to identify better lead compounds. Our results from this compound screen validated this multiplex screen assay as a useful method for identifying lead compounds that inhibit protein aggregation and/or reduce cytotoxicity caused by protein aggregates. In addition, this method might also be useful for studying the kinetic features of protein aggregate formation and effect of other cellular proteins on aggregates formations because of the robustness of assay performance and simple assay procedure.
In summary, we have demonstrated a multiplexed assay method which effectively measures cytotoxicity and protein aggregation in a cell-based model of Huntington’s disease. This assay method has been miniaturized into 1536-well plate format to screen large size compound collections for lead identification. This assay method also has the potential utility in screening other cell-based models of neurodegenerative diseases in which protein aggregation or plaque formation is implicated in the disease pathology [19 Bucciantini M, Giannoni E, Chiti F, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases Nature 2002; 416(6880): 507-11., 20 Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution J Mol Med 2003; 81(11): 678-99.].
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
This research was supported by the Molecular Libraries Initiative of the NIH Roadmap for Medical Research (5U54MH084681-02 and MH084839-01) and the Intramural Research Program of National Center for Advancing Translational Sciences, National Institutes of Health. We would like to thank E. Schweitzer for the PC12 cell lines and protocols, D. Leja for artwork, M. DeBernardi for confocal images, and W. Bowen for technical assistance with the Acumen Explorer protocol and manuscript.
ABBREVIATIONS
| HTT | =Huntingtin gene |
| HD | =Huntington’s Disease |
| HTS | =high throughput screening |
| LOPAC | =Library of Pharmacologically Active Compounds |
| MLPCN | =Molecular Libraries Probe Centers Network |