- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Arthritis Journal
(Discontinued)
ISSN: 1876-5394 ― Volume 7, 2014
Dendritic Cells in Autoimmune Diseases
Mohan S. Maddur1, 2, 3, Janakiraman Vani1, 2, 3, Jordan D. Dimitrov1, 2, 3, Kithiganahalli N. Balaji4, Sébastien Lacroix-Desmazes1, 2, 3, Srini V. Kaveri1, 2, 3, Jagadeesh Bayry*, 1, 2, 3
Abstract
Dendritic cells (DCs) are professional antigen presenting cells, which play a crucial role both in maintaining immune tolerance and in inducing adaptive immune responses. Therefore, DCs are the most sought in inciting autoimmunity and its sustenance to autoimmune diseases. The emerging knowledge of the importance of DCs, DC-derived cytokines, and intracellular signal transduction pathways in mediating autoimmune diseases and in creating an inflammatory environment provides a rationale for pursuing strategies to block these inflammatory pathways for therapeutic purposes.
Article Information
Identifiers and Pagination:
Year: 2010Volume: 3
First Page: 1
Last Page: 7
Publisher Id: TOARTHJ-3-1
DOI: 10.2174/1876539401003010001
Article History:
Received Date: 19/5/2009Revision Received Date: 14/9/2009
Acceptance Date: 14/9/2009
Electronic publication date: 12/1/2010
Collection year: 2010
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the INSERM U 872, Equipe 16, Centre de Recherche des Cordeliers, 15 rue de l’Ecole de Médicine, Paris, F-75006, France; Tel: 00 33 1 55 42 82 66; Fax: 00 33 1 55 42 82 62; E-mail: jagadeesh.bayry@crc.jussieu.fr
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 19-5-2009 |
Original Manuscript | Dendritic Cells in Autoimmune Diseases | |
Dendritic cells (DCs) are the prototype professional antigen presenting cells (APCs) derived from hemopoietic cells and are essential link between innate and adaptive arms of the immune system. They are sparse, but widely distributed in different types of tissues, serving as sentinels of the immune system [1]. DCs are capable of stimulating naïve T lymphocytes and hence play a central role in initiating primary immune responses [2]. In addition, DCs also play a critical role in maintaining immune tolerance through cross talk with T and B cells [1,3].
SUBSETS OF DENDRITIC CELLS
Despite similar basic function of antigen presentation, DCs differ in their developmental pathways, location, migratory properties, phenotypic markers and immunological functions. Thus, DCs are heterogeneous and several subtypes with distinct features have been identified in lymphoid and non-lymphoid tissues of human and mouse [1,3]. The major subtypes of human DCs are Langerhans cells, interstitial dermal DCs, monocyte-derived inflammatory DCs (Mo-DCs), BDCA-1+ and BDCA-3+ myeloid DC subsets, and plasmacytoid DCs (pDCs). The major subsets of mouse DCs include CD4-CD8αhiCD205hiCD11b- DCs, CD4+CD8α-CD205-CD11b+ DCs, CD4-CD8α-CD205-CD11b+ DCs, CD4-CD8α-CD205+CD11b- DCs, plasmacytoid DCs, and Langerhans DCs [3]. However, as depicted in Table 1, human and mouse DC subsets vary in their expression of CD markers (Table 1) [4].


MATURATION AND ACTIVATION OF DENDRITIC CELLS
Phenotypically, DCs are identified in “immature” and “mature” stage, based on the expression of surface markers [2]. Resting or immature DCs have high capacity to capture and internalize antigens due to surface expression of receptors that enables the recognition and endocytosis of potential antigens. These receptors include members of pattern recognition receptors (PRRs) family (such as Toll-like receptors (TLRs), C-type lectin receptors (CLRs), intracytoplasmic nucleotide oligomerization domain (NOD)-like receptors (NLRs)), Fc receptors (FcR), complement receptors, scavenger receptors and receptors involved in uptake of apoptotic bodies such as phosphatidylserine receptor. The expression of surface receptors differs with DC subsets and hence enables identification of subtypes [4]. DCs also possess numerous cytokine receptors that enable them to respond to the appropriate stimuli. Thus, immature DCs act as immunological sensors that receive stimuli from environment to alert the immune system. However, immature DCs are poor stimulators of T cells and must undergo a maturation and activation process.
Upon antigen capture by immature DCs, a complex process of maturation and activation is initiated based on the nature of the stimuli. The molecules that provide maturation-associated signals include pathogen-derived molecules (eg., dsRNA, LPS, CpG), dying cell components, tissue factors such as hyaluronan fragments, heparin sulphate, immune complexes and inflammatory cytokines [5]. The components of the dying cells include heat shock proteins (HSPs), high-mobility group box1 proteins (HMGB1), β-defensins, ATP and uric acid, which are collectively called damage-associated molecular pattern molecules (DAMPS) [6]. NF-κB and mitogen-activated protein kinase are the two major signaling pathways that are activated by these stimuli during maturation of DCs [7,8].
Maturation is defined as the series of phenotypic changes that enables DCs to initiate immunity as professional APCs [9]. This process involves similar morphological and functional changes in both human and mouse DCs [9] that include (i) loss of endocytic/phagocytic receptors; (ii) morphological changes such as the loss of adhesive structures, cytoskeleton reorganization, and the acquisition of high cellular motility; (iii) secretion of immunoregulatory cytokines and chemokines such as IL-1β, IL-6, IL-10, IL-12, IL-23, TNF, CCL17, CCL19 and CCL22; (iv) up-regulation of co-stimulatory and adhesion molecules CD80, CD86, CD40, CD54, CD58 critical in mediating clustering with and activation of T cells; (v) translocation of MHC class I/II compartments to the cell surface; and (vi) migration of mature DCs into regional/draining lymphoid tissue. As a result, antigen acquired in peripheral sites is retained, processed and presented to T cells in lymph nodes.
INDUCTION OF ADAPTIVE IMMUNE RESPONSES BY DENDRITIC CELLS
The process of T-cell activation is regulated primarily by signaling events derived from the T-cell receptor TCR (signal 1) and from co-stimulatory and adhesion molecules (signal 2) CD80, CD86, CD40, CD54, CD58 on APCs and CD28, CD154, CD11a and CD2 on T cells. The CD80 and/or CD86–CD28 interactions provide signals important for T-cell activation and survival. CD40–CD154 interactions are crucial for the development of CD4+ T-dependent effector functions such as help for B-cell differentiation and class switch. The interactions of adhesion molecules CD54 and CD11a, and CD58 and CD2 play a central part in the clustering of DCs and CD4+ T lymphocytes and facilitate TCR triggering by increasing T-cell–DC interactions at low antigen densities. An additional signal has also been proposed in the form of immunomodulatory cytokines (signal 3) produced by activated DCs that dictate the polarization of CD4+ T cells towards Th1, Th2, Th17 or regulatory T cells (Tregs) (Fig. 1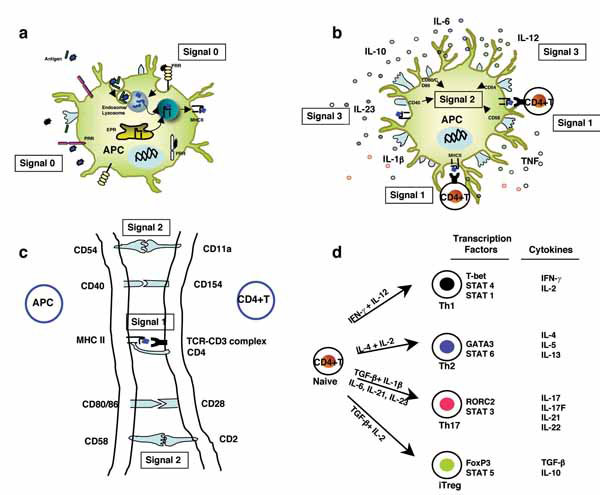 ) [10]. Interleukin (IL)-12 has an important role in the differentiation of Th1 cells, and IL-4 is crucial for Th2 differentiation. IL-1β and IL-6 promote IL-17 and IL-21 production from activated and memory human T cells. IL-21 promotes the differentiation of human Th17 cells from naïve CD4+ T cells in the presence of transforming growth factor (TGF)-β, whereas IL-23 might further expand or stabilize differentiated Th17 cells [11-14].
) [10]. Interleukin (IL)-12 has an important role in the differentiation of Th1 cells, and IL-4 is crucial for Th2 differentiation. IL-1β and IL-6 promote IL-17 and IL-21 production from activated and memory human T cells. IL-21 promotes the differentiation of human Th17 cells from naïve CD4+ T cells in the presence of transforming growth factor (TGF)-β, whereas IL-23 might further expand or stabilize differentiated Th17 cells [11-14].

In addition to T cell stimulation, DCs contribute to the stimulation of B cells, both in the lymph node T cell areas and in germinal centers. Antigens in the immune complex captured by inhibitory Fc receptor (FcRγIIB) can be retained in non-degradative intracellular vesicles and presented in native form to B cells [15]. DCs can also modulate the functions of non-specific effectors such as natural killer cells and natural killer T cells and influence the outcome of immune response [16, 17].
DENDRITIC CELLS IN IMMUNE TOLERANCE: GENERATION OF REGULATORY T CELLS
In addition to their central role in initiating adaptive immunity, DCs are also involved in the induction of immune tolerance in both central and peripheral compartments by different regulatory mechanisms that include, clonal deletion, clonal anergy, receptor editing of T/B cells and generation of Tregs [16, 17]. Role of DCs in generation of regulatory T cells is of prime importance [15]. In human thymus, the thymic stromal lymhopoietin (TSLP) derived from the hassall’s corpuscles instruct DCs to switch from negative selection of autoreactive T cells to positive selection of natural Tregs (nTregs). Interestingly, the preformed immature myeloid tolerogenic DCs carrying peripheral antigens migrate into the thymus and induce the generation of nTregs in vivo [15]. In addition, immature/tolerogenic DCs or DCs stimulated to produce IL-10/ TGF-β induce Tregs in peripheral tissues referred to as induced regulatory cells (iTregs) [18, 19]. Tregs play a crucial role in maintaining immune tolerance by acting on several cellular targets [20, 21].
DENDRITIC CELLS: INCITING AND SUSTAINING AUTOIMMUNITY
Autoimmunity refers to inappropriate immune response against self-components of the host that might result in pathological conditions. A low level of autoreactivity is acknowledged as physiologic and crucial for normal immune function [22]. Autoimmune diseases are clinical syndromes that occur in selected genetic backgrounds characterised by the abnormal activation of T cells, B cells or both in the absence of on going infection or other discernible cause [23]. Autoimmune diseases affect approximately 5% of the population based on genetic characters and environmental factors. They can be clinically classified either systemic (eg., systemic lupus erythematosus (SLE) or organ specific (eg., type 1 diabetes mellitus) [23].
A breakdown in immune tolerance is central to the initiation and progression of an autoimmune disease. Given the central role in induction of immune response and tolerance, DCs are the most sought after cells in inciting autoimmunity and its sustenance to autoimmune diseases [18, 24-28]. In human leukocyte antigen-restricted autoimmune diseases, DC priming of the immune response in lymphoid organs is a requisite first step that leads to the activation of T cells and the production of pathogenic autoantibodies and the ensuing chronic inflammatory process [29-31]. Individuals with autoimmune disease show a high number of aberrantly activated DCs either in circulation or in the autoimmune lesions secreting large amounts of proinflammatory cytokines that mediate inflammation and differentiation of pathogenic Th1 and Th17 cells [26]. In the mouse model, altered DC homeostasis has been demonstrated to result in an apparent autoimmune response, thus signifying a central role of DCs in maintaining immune self-tolerance and potential to incite autoimmunity [32, 33]. Further, defects in the apoptosis of the immune cells and defective clearance of apoptotic cell debris due to an impaired uptake can result in development of autoimmunity. The priming of autoreactive lymphocytes by activated DCs that have taken up apoptotic materials may lead to break down of self-tolerance [34]. The role of TLRs expressed on different DC subsets has been increasingly evidenced in the pathogenesis of many autoimmune diseases [35, 36]. An important sequel of continued antigenic stimulation via DCs is the formation of lymphoid structures at the site of inflammation. By coordinating the recruitment and/or activation of other immune cells, DCs can drive the generation of ectopic lymphoid tissues, as in the case of inflamed synovia in rheumatoid arthritis (RA) and SLE [18, 19].
Although Tregs can suppress the maturation and activation and the production of inflammatory cytokines by recently activated DCs (Fig. 2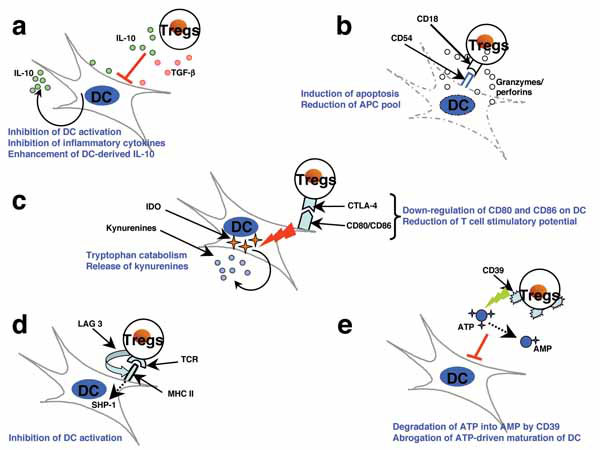 ) [31, 37-40], chronically activated DCs can escape suppression by Tregs and generate activated T cells that are refractory to the suppression by Tregs. High amounts of pro-inflammatory cytokines that are released by DCs due to constant and prolonged activation can render Tregs functionally defective. For example, prolonged exposure to TNF can inhibit the expression of FoxP3 and the function of Tregs by signaling through TNF receptor II [41]. Further, IL-6, IL-12, and IL-1β may also promote autoimmunity by interfering with the regulatory functions of Tregs and promoting activation and expansion of effector T cells [31]. In addition to directly suppressing the functions of Tregs, DC-derived cytokines such as IL-6, IL-1β, and IL-23 can reciprocally regulate the populations of iTregs and IL-17-producing pathogenic Th17 cells [42].
) [31, 37-40], chronically activated DCs can escape suppression by Tregs and generate activated T cells that are refractory to the suppression by Tregs. High amounts of pro-inflammatory cytokines that are released by DCs due to constant and prolonged activation can render Tregs functionally defective. For example, prolonged exposure to TNF can inhibit the expression of FoxP3 and the function of Tregs by signaling through TNF receptor II [41]. Further, IL-6, IL-12, and IL-1β may also promote autoimmunity by interfering with the regulatory functions of Tregs and promoting activation and expansion of effector T cells [31]. In addition to directly suppressing the functions of Tregs, DC-derived cytokines such as IL-6, IL-1β, and IL-23 can reciprocally regulate the populations of iTregs and IL-17-producing pathogenic Th17 cells [42].

Type 1 interferons (IFN-α and IFN-β) are secreted in large amount by pDCs not only following viral infection, but also in autoimmune diseases [1]. In psoriasis, pDCs accumulate in the inflamed skin at an early stage of the disease, and these cells secrete type I IFN [43]. In insulin-dependent diabetes mellitus, elevated expression of IFN was found in the pancreas of recently diagnosed patients, though the role of pDCs is unclear [44]. In idiopathic inflammatory myopathies, DCs present in inclusion body myositis and polymyositis are primarily myeloid DCs, whereas in dermatomyositis, type I IFN producing pDCs have been found abundantly in muscle tissue [45]. In Sjogren’s syndrome, an autoimmune disease affecting mainly salivary and lacrimal glands, pDCs infiltrate salivary glands, where IFN-inducible genes are over-expressed [46]. In immune thrombocytopenic pupura patients, DCs are found to be highly efficient in presenting apoptotic platelets and stimulating autologous T-cell proliferation [47].
DENDRITIC CELLS IN AUTOIMMUNE DISEASES: FEW SPECIFIC EXAMPLES
Rheumatoid Arthritis (RA)
Spontaneous arthritis animal models resembling RA have revealed defects related to interaction of DCs and thymocytes and as a consequence escape of pathogenic autoreactive T cells. These T cells upon stimulation by activated DCs in the presence of TNF-α and IL-1 are shown to drive autoantibody production and proinflammatory arthritogenic response [48]. Further, DCs can present altered citrullinated collagens and fibrin to autoreactive T cells (Table 2) [49]. DCs enter the inflamed synovium through blood vessels and are found to have activated NF-κB pathway. Mature myeloid DCs are in perivascular T cell rich areas of synovial tissue and immature DCs are abundant in the synovial lining and sublining layers of synovium [28, 49]. Myeloid DCs can enhance inflammation in the synovium by secreting IL-23 that induces Th17 cells. pDCs are also observed in the inflamed synovium that might respond to immunostimulatory nucleic acid sequences complexed to rheumatoid factor, by producing IFN-α [50]. TNF-α is a dominant inflammatory cytokine that is over-produced in the joints by macrophages and mature myeloid DCs [51].
Systemic Lupus Erythematosus (SLE)
IFN-α is the cytokine implicated in pathogenesis of SLE. pDCs are the main source of IFN-α, produced in response to nucleic acid-containing immune complexes which are detected by TLR7 and TLR9 (Table 2) [52]. Unabated induction of DC maturation by IFN-α has been proposed to drive the autoimmune responses in systemic SLE [53]. Thus, IFN-α induces differentiation and activation of DCs from monocytes in SLE individuals, which can activate autoreactive T and B cells [53, 54]. IFN-α/β also enhances B-cell response to nucleic acid-containing immune complexes by upregulating TLR7 and promoting cell death to release increased amounts of nucleic acid. pDC-produced IFNs initiate a self-perpetuating feedback loop to drive autoantibody production in SLE [52]. Aberrant phenotype and function of DCs in SLE is also observed [55-57].
Multiple Sclerosis (MS)
MS is a T cell-mediated disease, where Th1 and Th17 cells predominate resulting in chronic inflammation, demyelination and progressive paralysis [58]. DCs are recruited to MS lesions, where uptake and presentation of self-antigens which are made available by continuous myelin destruction, may contribute to the local activation and expansion of presumably pathogenic T cells (Table 2) [59, 60]. DCs found within MS lesions have been shown to be functionally abnormal [61]. Further, intra-cerebral DCs were found to critically modulate encephalitogenic versus regulatory immune responses in CNS of EAE mice, an animal model of MS and hence considered to be rate-limiting factor for neuro-inflammation [62].
CONCLUSION
DCs are the prime initiators and mediators of autoimmune diseases in an altered host microenvironment (cytokine imbalance) arising out of environmental and genetic perturbations. DCs act in concert with adaptive and other innate immune cells in moulding the autoimmune response. The emerging knowledge of the importance of DCs, DC-derived cytokines, and intracellular signal transduction pathways in mediating and creating an inflammatory environment in autoimmune diseases provides a rationale for pursuing strategies to block these inflammatory pathways for the therapeutic purposes.
ACKNOWLEDGEMENTS
Supported by grants Institut National de la Santé et de la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS), Université Paris Descartes-Paris 5, Université Pierre et Marie Curie-Paris 6, Agence Nationale de la Recherche (ANR-07-JCJC-0100-01), Coopération Institut National de la Santé et de la Recherche Médicale–Indian Council for Medical Research (INSERM-ICMR-AO 2009/2010, eSPIN (European Scientific Progress - Immunoglobulins in Neurology) award 2009 and Talents research grant from Talecris Biotherapeutics.
REFERENCES
Endorsements
Browse Contents
Table of Contents
- SUBSETS OF DENDRITIC CELLS
- MATURATION AND ACTIVATION OF DENDRITIC CELLS
- INDUCTION OF ADAPTIVE IMMUNE RESPONSES BY DENDRITIC CELLS
- DENDRITIC CELLS IN IMMUNE TOLERANCE: GENERATION OF REGULATORY T CELLS
- DENDRITIC CELLS: INCITING AND SUSTAINING AUTOIMMUNITY
- DENDRITIC CELLS IN AUTOIMMUNE DISEASES: FEW SPECIFIC EXAMPLES
- CONCLUSION