- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Structural Chemistry of Human SET Domain Protein Methyltransferases
Matthieu Schapira*, 1, 2
Abstract
There are about fifty SET domain protein methyltransferases (PMTs) in the human genome, that transfer a methyl group from S-adenosyl-L-methionine (SAM) to substrate lysines on histone tails or other peptides. A number of structures in complex with cofactor, substrate, or inhibitors revealed the mechanisms of substrate recognition, methylation state specificity, and chemical inhibition. Based on these structures, we review the structural chemistry of SET domain PMTs, and propose general concepts towards the development of selective inhibitors.
Article Information
Identifiers and Pagination:
Year: 2011Volume: 5
Issue: Suppl 1
First Page: 85
Last Page: 94
Publisher Id: CCGTM-5-85
DOI: 10.2174/1875397301005010085
Article History:
Received Date: 27/1/2011Revision Received Date: 6/4/2011
Acceptance Date: 25/4/2011
Electronic publication date: 22/8/2011
Collection year: 2011
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Structural Genomics Consortium, University of Toronto, MaRS Centre, Toronto, Ontario, M5G 1L7, Canada; Tel: 416-978-3092; Fax: 416-946-0880; E-mail: matthieu.schapira@utoronto.ca
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 27-1-2011 |
Original Manuscript | Structural Chemistry of Human SET Domain Protein Methyltransferases | |
INTRODUCTION
Epigenetics mechanisms rely extensively on histone-mediated signaling, in which chemical modifications can make or break complex biological circuits [1Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond Nature 2003; 425: 475-9., 2Kouzarides T. Chromatin modifications and their function Cell 2007; 128: 693-705.]. Among the different histone marks, methylation of specific lysine and arginine side-chains can regulate chromatin compaction, repress or activate transcription, and control cellular differentiation [3Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery Nat Rev Drug Discov 2009; 8: 724-32., 4Martin C, Zhang Y. The diverse functions of histone lysine methylation Nat Rev Mol Cell Biol 2005; 6: 838-49.]. The transfer of a methyl group from the cofactor S-adenosyl-L-methione (SAM) to substrate peptides can be catalyzed by two classes of enzymes [5Schubert HL, Blumenthal RM, Cheng X. Many paths to methyltransfer: a chronicle of convergence Trends Biochem Sci 2003; 28: 329-5., 6Petrossian TC, Clarke SG. Uncovering the human methyltransferasome Mol Cell Proteomics 2011; 10: M110-000976.]. Nine arginine protein methyltransferases (PMTs) are known in human, whose function, structure, chemistry, and chemical inhibition have recently been reviewed [7Di Lorenzo A, Bedford MT. Histone arginine methylation FEBS Lett 2010. [Epub ahead of print]-9Spannhoff A, Hauser AT, Heinke R, Sippl W, Jung M. The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors ChemMedChem 2009; 4: 1568-82.] (Yost et al. this issue). Lysine methylation is catalyzed by SET domain PMTs, a family of about fifty proteins in human [10Volkel P, Angrand PO. The control of histone lysine methylation in epigenetic regulation Biochimie 2007; 89: 1-20.], and DOT1L, an enzyme that lacks the canonical SET domain, but shares the same fold as arginine PMTs [11Min J, Feng Q, Li Z, Zhang Y, Xu RM. Structure of the catalytic domain of human DOT1L. a non-SET domain nucleosomal histone methyltransferase Cell 2003; 112: 711-23.]. This review focuses on the SET domain lysine PMTs. The SET domain is a sequence of 130 amino-acids, originally named after the Drosophila genes Su(var), E(z) and Trithorax in which it was originally identified. It is defined by a specific fold organized around a pseudo-knot, and by the presence of two signature motifs, ELxF/YDY and NHS/CxxPN, x being any amino-acid [12Dillon SC, Zhang X, Trievel RC, Cheng X. The SET-domain protein superfamily: protein lysine methyltransferases Genome Biol 2005; 6: 227.-14Xiao B, Jing C, Wilson JR, et al. Structure and catalytic mechanism of the human histone methyltransferase SET7/9 Nature 2003; 421: 652-.].
While the SET domain is responsible for catalysis, the methyltransferase activity of PMTs also depends on the presence of adjacent domains that recruit the substrate, or other structural modules, sometimes remote, that act as binding platforms for interaction partners within large multi-subunit complexes [10Volkel P, Angrand PO. The control of histone lysine methylation in epigenetic regulation Biochimie 2007; 89: 1-20.]. For instance, the PMT EZH2 is only active within the PRC2 complex when associated with EED and SUZ12; recruitment of EED is mediated by a region located 500 residues upstream of EZH2’s SET domain [15Han Z, Xing X, Hu M, Zhang Y, Liu P, Chai J. Structural basis of EZH2 recognition by EED Structure 2007; 15: 1306-5.]. Remote structural modules may not be necessary for PMT activity, but sometimes recognize the methylation substrate or reaction product. For instance, it was shown that an Ankyrin repeat distinct from the catalytic domain of GLP could recognize mono- or di-methylated lysine 9 of histone 3, the very reaction product of GLP’s SET domain [16Collins RE, Northrop JP, Horton JR, et al. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules Nat Struct Mol Biol 2008; 15: 245-50.].
As previously observed for histone deacetylases and histone acetyltransferases, it is becoming clear that histones are not the only subtrates of some PMTs. For instance, G9a and GLP can methylate the tumor suppressor p53 [17Huang J, Dorsey J, Chuikov S, et al. G9a and Glp methylate lysine 373 in the tumor suppressor p53 J Biol Chem 2010; 285: 9636-41.]. These emerging signaling mechanisms, unrelated to the histone code, add to the already large body of evidence associating SET domain PMTs to multiple disease areas, and further drive the research community towards the development of chemical tools to better interrogate their function [3Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery Nat Rev Drug Discov 2009; 8: 724-32.].
OVERALL ARCHITECTURE OF THE CATALYTIC DOMAIN
The catalytic domain is composed of a core SET domain that is structurally conserved and includes residues critical for catalysis, surrounded with a limited set of regions that vary in nature, sequence and shape (Fig. 2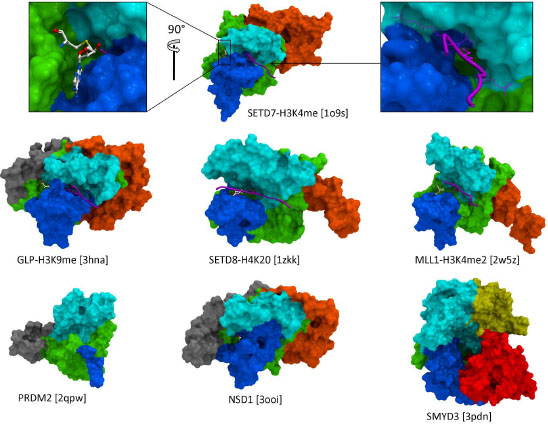 ). These adjacent domains act like a shell around the SET fold, and can be divided into two categories. First, the I-SET and post-SET domains (respectively inserted within, and immediately C-terminal to the SET fold) form the binding groove for the substrate peptide, and, to a lesser extent, contribute to the cofactor binding pocket. A landmark feature of SET domain PMTs is that the substrate peptide and cofactor bind distinct sites, on different sides of the protein, and meet at the core of the structure where catalysis takes place. Available ternary complexes of SETD7 and GLP reveal how the side-chain of the substrate lysine inserts into a narrow channel at the junction of the SET, Post-SET, and I-SET domains [14Xiao B, Jing C, Wilson JR, et al. Structure and catalytic mechanism of the human histone methyltransferase SET7/9 Nature 2003; 421: 652-., 18Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570.]. In this configuration, the lysine is shielded from the solvent, which is believed to be required for catalysis [8Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications Biochim Biophys Acta 2009; 1789: 45-57.]. In the SETD8 ternary structure, a wide pocket, rather than a channel, is occupied by the histone lysine and a flanking histidine [19Couture JF, Collazo E, Brunzelle JS, Trievel RC. Structural and functional analysis of SET8. a histone H4 Lys-20 methyltransferase Genes Dev 2005; 19: 1455-65., 20Xiao B, Jing C, Kelly G, et al. Specificity and mechanism of the histone methyltransferase Pr-Set7 Genes Dev 2005; 19: 1444-54.]. A catalytically inactive structure of MLL1 features a more open peptide binding groove, which leaves the substrate lysine exposed to solvent. Other protein binding partners, part of the MLL complex, are expected to stabilize the active conformation, probably closer to that captured by the GLP, SETD7 and SETD8 structures [21Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks Mol Cell 2009; 33: 181-91.]. Since both I- and Post-SET domains are involved in substrate recognition, it comes as no surprise that both are present in all SET domain PMTs. The I-SET domain has a fixed topology, and is structurally static, while the post-SET domain adopts variable folds and is structurally dynamic, which has implications for the mechanism of substrate recognition (vide infra). Available PRDM structures reveal an extremely short and unfolded post-SET, which may explain the absence of observed biochemical activity for these protein constructs.
). These adjacent domains act like a shell around the SET fold, and can be divided into two categories. First, the I-SET and post-SET domains (respectively inserted within, and immediately C-terminal to the SET fold) form the binding groove for the substrate peptide, and, to a lesser extent, contribute to the cofactor binding pocket. A landmark feature of SET domain PMTs is that the substrate peptide and cofactor bind distinct sites, on different sides of the protein, and meet at the core of the structure where catalysis takes place. Available ternary complexes of SETD7 and GLP reveal how the side-chain of the substrate lysine inserts into a narrow channel at the junction of the SET, Post-SET, and I-SET domains [14Xiao B, Jing C, Wilson JR, et al. Structure and catalytic mechanism of the human histone methyltransferase SET7/9 Nature 2003; 421: 652-., 18Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570.]. In this configuration, the lysine is shielded from the solvent, which is believed to be required for catalysis [8Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications Biochim Biophys Acta 2009; 1789: 45-57.]. In the SETD8 ternary structure, a wide pocket, rather than a channel, is occupied by the histone lysine and a flanking histidine [19Couture JF, Collazo E, Brunzelle JS, Trievel RC. Structural and functional analysis of SET8. a histone H4 Lys-20 methyltransferase Genes Dev 2005; 19: 1455-65., 20Xiao B, Jing C, Kelly G, et al. Specificity and mechanism of the histone methyltransferase Pr-Set7 Genes Dev 2005; 19: 1444-54.]. A catalytically inactive structure of MLL1 features a more open peptide binding groove, which leaves the substrate lysine exposed to solvent. Other protein binding partners, part of the MLL complex, are expected to stabilize the active conformation, probably closer to that captured by the GLP, SETD7 and SETD8 structures [21Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks Mol Cell 2009; 33: 181-91.]. Since both I- and Post-SET domains are involved in substrate recognition, it comes as no surprise that both are present in all SET domain PMTs. The I-SET domain has a fixed topology, and is structurally static, while the post-SET domain adopts variable folds and is structurally dynamic, which has implications for the mechanism of substrate recognition (vide infra). Available PRDM structures reveal an extremely short and unfolded post-SET, which may explain the absence of observed biochemical activity for these protein constructs.

Other domains surrounding the SET domain include Pre-SET (N-terminal to SET), N-SET (N-terminal to SET or Pre-SET), MYND (between SET and I-SET), and CTD (C-terminal to Post-SET) (Fig. 2). It is believed that some, if not all of these variable domains are acting as binding interfaces to other proteins or DNA. A general concept would therefore be that different combinations of domains with diverse sequence, structure, and electrostatics, would dress the core SET fold in very distinct ways, and allow selective recruitment of interaction partners, or facilitate specific positioning relative to the nucleosome, with functional implications.
Recent structures of SMYD proteins illustrate how the CTD domain can adopt an open, catalytically competent conformation, as observed in SMYD1, or an inactive conformation that partially occupies the substrate peptide binding site (Fig. 3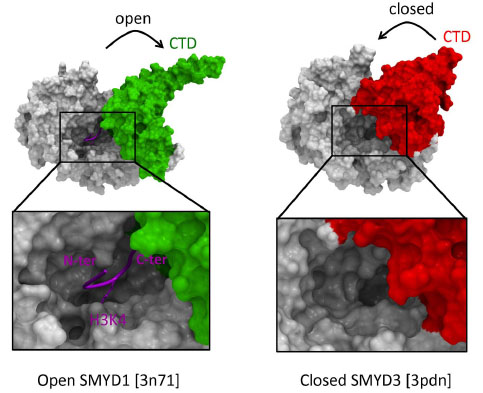 ) [22Sirinupong N, Brunzelle J, Doko E, Yang Z. Structural insights into the autoinhibition and posttranslational activation of histone methyltransferase SmyD3 J Mol Biol 2011; 406(1
): 149-59., 23Sirinupong N, Brunzelle J, Ye J, Pirzada A, Nico L, Yang Z. Crystal structure of cardiac-specific histone methyltransferase SmyD1 reveals unusual active site architecture J Biol Chem 2010; 285: 40635-4.]. It was proposed that binding of HSP90, which activates SMYD3, stabilizes the open conformation of the protein. Domains adjacent to SET may therefore not only act as protein interaction interfaces, but also as auto-inhibitory components.
) [22Sirinupong N, Brunzelle J, Doko E, Yang Z. Structural insights into the autoinhibition and posttranslational activation of histone methyltransferase SmyD3 J Mol Biol 2011; 406(1
): 149-59., 23Sirinupong N, Brunzelle J, Ye J, Pirzada A, Nico L, Yang Z. Crystal structure of cardiac-specific histone methyltransferase SmyD1 reveals unusual active site architecture J Biol Chem 2010; 285: 40635-4.]. It was proposed that binding of HSP90, which activates SMYD3, stabilizes the open conformation of the protein. Domains adjacent to SET may therefore not only act as protein interaction interfaces, but also as auto-inhibitory components.

SUBSTRATE RECOGNITION
Display of the molecular surface of PMTs according to their electrostatic potential reveals that the substrate-binding groove is consistently electronegative, as illustrated Fig. (4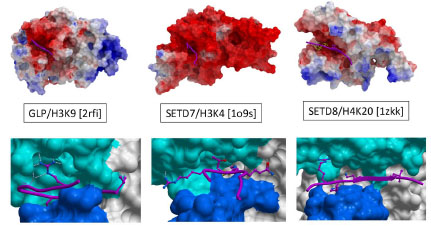 ) for an H3K9, H3K4, and H4K20 PMT (GLP, SETD7 and SETD8 respectively). This is in contrast with histone tails, which are enriched in lysine and arginine residues, and highly electropositive. This observation suggests a general mechanism whereby long-range electrostatic attractions can bring the PMTs and their peptide substrates together in a loose complex, prior to sequence-specific recognition.
) for an H3K9, H3K4, and H4K20 PMT (GLP, SETD7 and SETD8 respectively). This is in contrast with histone tails, which are enriched in lysine and arginine residues, and highly electropositive. This observation suggests a general mechanism whereby long-range electrostatic attractions can bring the PMTs and their peptide substrates together in a loose complex, prior to sequence-specific recognition.

A close inspection of PMT structures co-crystallized with substrate peptides reveals that the substrate lysine is anchored in a deep channel, and is the major contributor to binding enthalpy. Surprisingly, in all available structures, an arginine side-chain located one to four residues upstream or downstream the substrate lysine is the next most important contributor to interaction, and makes extensive contacts with a well-defined cleft of the I-SET domain (Fig. 4) [14Xiao B, Jing C, Wilson JR, et al. Structure and catalytic mechanism of the human histone methyltransferase SET7/9 Nature 2003; 421: 652-., 18Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570., 19Couture JF, Collazo E, Brunzelle JS, Trievel RC. Structural and functional analysis of SET8. a histone H4 Lys-20 methyltransferase Genes Dev 2005; 19: 1455-65., 21Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks Mol Cell 2009; 33: 181-91., 24Wang M, Mok MW, Harper H, et al. Structural genomics of histone tail recognition Bioinformatics 2010; 26: 2629-30.]. Interestingly, the shape, structural environment, and position of this cleft relative to the lysine binding channel varies from one enzyme to the other, suggesting that it could be exploited to design selective inhibitors. This concept was validated in the case of G9a and GLP. Indeed, co-crystallized selective inhibitors were shown to occupy the arginine binding site, as discussed below [25Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of
histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3., 26Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7.]. Another observation with possible mechanistic consequences is the fact that histone residues projecting towards the groove are enriched in serine and threonine, two other sites of post-translational modification. It is tempting to speculate that this trend reflects a general structural mechanism where distinct combinations of histone marks would antagonize or possibly enhance substrate recognition by specific PMTs. This hypothesis is supported by some experimental observations, but is beyond the scope of this study (see for instance [27Couture JF, Collazo E, Hauk G, Trievel RC. Structural basis for the methylation site specificity of SET7/9 Nat Struct Mol Biol 2006; 13: 140-6.-29Binda O, LeRoy G, Bua DJ, Garcia BA, Gozani O. Trimethylation of histone H3 lysine 4 impairs methylation of histone H3 lysine 9: regulation of lysine methyltransferases by
physical interaction with their substrates Epigenetics 2010; 5: 767-5.]).
As mentioned above, the I-SET domain varies in sequence, but is structurally conserved across PMTs. On the other hand, the Post-SET domain has variable topologies, sometimes organized around a coordinating Zn atom, as is observed for instance in the H3K9 PMTs G9a [18Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570.], or the H3K4 PMT MLL1 [21Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks Mol Cell 2009; 33: 181-91.]. SETD7 was crystallized in its apo state, in a binary complex with cofactor, and ternary complex with cofactor and substrate peptide [14Xiao B, Jing C, Wilson JR, et al. Structure and catalytic mechanism of the human histone methyltransferase SET7/9 Nature 2003; 421: 652-., 30Kwon T, Chang JH, Kwak E, et al. Mechanism of histone lysine methyl transfer revealed by the structure of SET7/9-AdoMet EMBO J 2003; 22: 292-303., 31Wilson JR, Jing C, Walker PA, et al. Crystal structure and functional analysis of the histone methyltransferase SET7/9 Cell 2002; 111: 105-5.]. The I-SET structure remains unchanged between the three states (with the exception of a tryptophan side-chain), while the conformation of the Post-SET domain varies considerably (Fig. 5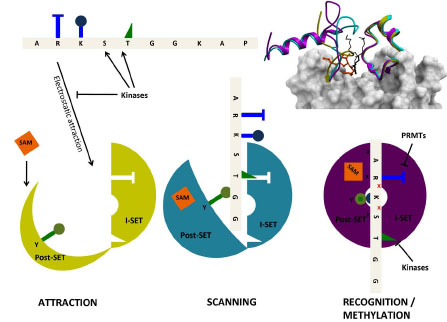 ). Interestingly, a sequential mechanism seems to take place: the apo-conformation is completely unfolded. Binding of the cofactor induces partial folding, where an helix contributing to the cofactor binding site adopts its final conformation. Finally, proper positioning of the substrate peptide relative to the static I-SET induces a final conformational adjustment of the Post-SET domain. Based on similar observations, a model was proposed for the processivity of substrate methylation where an opening and closing motion of the Post-SET domain would allow release into the solvent of the cofactor and of a proton from the substrate lysine after a first methylation event. Cofactor exchange and deprotonation of the substrate are both necessary before further methylation can take place [20Xiao B, Jing C, Kelly G, et al. Specificity and mechanism of the histone methyltransferase Pr-Set7 Genes Dev 2005; 19: 1444-54.].
). Interestingly, a sequential mechanism seems to take place: the apo-conformation is completely unfolded. Binding of the cofactor induces partial folding, where an helix contributing to the cofactor binding site adopts its final conformation. Finally, proper positioning of the substrate peptide relative to the static I-SET induces a final conformational adjustment of the Post-SET domain. Based on similar observations, a model was proposed for the processivity of substrate methylation where an opening and closing motion of the Post-SET domain would allow release into the solvent of the cofactor and of a proton from the substrate lysine after a first methylation event. Cofactor exchange and deprotonation of the substrate are both necessary before further methylation can take place [20Xiao B, Jing C, Kelly G, et al. Specificity and mechanism of the histone methyltransferase Pr-Set7 Genes Dev 2005; 19: 1444-54.].

We propose a general structural mechanism integrating electrostatic phenomena, Post-SET dynamics, and histone mark cross-talk (Fig. 5). Long range electrostatic attractions bring together the electropositive histone tail and a loose electronegative binding groove, composed of a pre-formed I-SET and open Post-SET. SAM binding stabilizes a partially folded Post-SET conformation. I-SET acts as a reading platform for the substrate peptide. The PMT may slide along the histone tail, held in place by non-specific electrostatics. Once a specific combination of histone side-chains comes into register with I-SET, the substrate lysine loses a proton to the solvent, and the complex clicks into a catalytically competent conformation where (1) a catalytic tyrsosine located at the C-terminus of the SET domain completes formation of the lysine channel and projects towards the active site, (2) a conserved double-hydrogen bond flanking the substrate lysine is engaged with the I-SET domain, (3) the post-SET domain closes onto the bound peptide, shielding the catalytic center from solvent. Histone marks deposited by other enzymes on flanking serine, threonine or arginine side-chains can affect the formation of this catalytically competent state.
Structures of the three H3K36 PMTs SETD2, SETMAR, and NSD1 (PDB codes 3h6l, 3bo5, 3ooi resp.), and the H3K9 tri-methylase SUV39H2 (PDB code 2r3a) are lacking a peptide binding groove, which seems to contradict this model (Fig. 6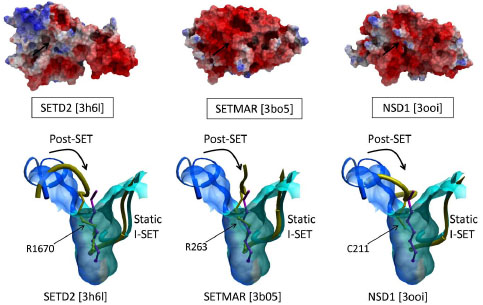 , top). In these structures, the I-SET domain superimposes well with the I-SET of active structures, such as histone-bound GLP, but a side-chain of the Post-SET domain projects into what would be the substrate lysine channel, and flanking Post-SET residues occupy the peptide binding groove (Fig. 6, bottom). The functional relevance of this auto-inhibitory mechanism, originally reported for SUV39H2, remains unknown at this time [18Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570.].
, top). In these structures, the I-SET domain superimposes well with the I-SET of active structures, such as histone-bound GLP, but a side-chain of the Post-SET domain projects into what would be the substrate lysine channel, and flanking Post-SET residues occupy the peptide binding groove (Fig. 6, bottom). The functional relevance of this auto-inhibitory mechanism, originally reported for SUV39H2, remains unknown at this time [18Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570.].

CATALYSIS
Catalysis takes place at the SET domain, where the departing methyl of the cofactor lies in close proximity with the de-protonated ε-nitrogen of the substrate lysine, at the bottom of the lysine channel (Fig. 7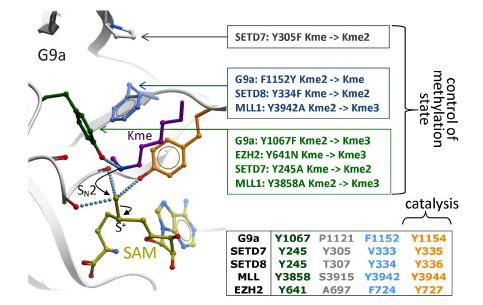 ). The nucleophilicity of the departing methyl group is enhanced by neighboring main chain carbonyl oxygens, and the hydroxyl end of a catalytic tyrosine. Another surrounding tyrosine forms a hydrogen bond with the substrate lysine, thereby aligning the lone pair of the deprotonated ε-nitrogen with the scissile methyl-sulfur bond. A nucleophilic attack follows, which results in methylation of the lysine, and release of SAH [8Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications Biochim Biophys Acta 2009; 1789: 45-57.]. A correlation has been observed between the number of residues susceptible of forming a hydrogen bond with the substrate lysine - generally a tyrosine - and the methylation state. Indeed, adding hydrogen bonds restrains the rotational freedom of the nitrogen atom, which is necessary to align its lone pair with the scissile bond of the sulfonium group. Mutational analyses have confirmed experimentally that a Tyr-Phe switch in the active site can effectively control the methylation product (Fig. 7) [18Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570., 32Guo HB, Guo H. Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity Proc Natl Acad Sci U S A 2007; 104: 8797-02.-34Couture JF, Dirk LM, Brunzelle JS, Houtz RL, Trievel RC. Structural origins for the product specificity of SET domain protein methyltransferases Proc Natl Acad Sci U S A 2008; 105: 20659-64.]. Additionally, the extra bulk created by the tyrosine’s hydroxy group, or, as shown in SETD8, by a bound water molecule, can sterically prohibit higher methylation states [34Couture JF, Dirk LM, Brunzelle JS, Houtz RL, Trievel RC. Structural origins for the product specificity of SET domain protein methyltransferases Proc Natl Acad Sci U S A 2008; 105: 20659-64.]. Interestingly, this switch was recently reported as a frequent somatic mutation in lymphoma, changing the EZH2 from a multifunctional mono- di- and trimethylase to an enzyne with increased trimethylase activity, but little or no mono- and dimethylase activity [35Yap DB, Chu J, Berg T, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation Blood 2011; 117(8
): 2451-9., 36Sneeringer CJ, Scott MP, Kuntz KW, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas Proc Natl Acad Sci U S A 2010; 107: 20980-5.]. Inhibitors specifically recognizing the mutant enzyme may be of interest.
). The nucleophilicity of the departing methyl group is enhanced by neighboring main chain carbonyl oxygens, and the hydroxyl end of a catalytic tyrosine. Another surrounding tyrosine forms a hydrogen bond with the substrate lysine, thereby aligning the lone pair of the deprotonated ε-nitrogen with the scissile methyl-sulfur bond. A nucleophilic attack follows, which results in methylation of the lysine, and release of SAH [8Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications Biochim Biophys Acta 2009; 1789: 45-57.]. A correlation has been observed between the number of residues susceptible of forming a hydrogen bond with the substrate lysine - generally a tyrosine - and the methylation state. Indeed, adding hydrogen bonds restrains the rotational freedom of the nitrogen atom, which is necessary to align its lone pair with the scissile bond of the sulfonium group. Mutational analyses have confirmed experimentally that a Tyr-Phe switch in the active site can effectively control the methylation product (Fig. 7) [18Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570., 32Guo HB, Guo H. Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity Proc Natl Acad Sci U S A 2007; 104: 8797-02.-34Couture JF, Dirk LM, Brunzelle JS, Houtz RL, Trievel RC. Structural origins for the product specificity of SET domain protein methyltransferases Proc Natl Acad Sci U S A 2008; 105: 20659-64.]. Additionally, the extra bulk created by the tyrosine’s hydroxy group, or, as shown in SETD8, by a bound water molecule, can sterically prohibit higher methylation states [34Couture JF, Dirk LM, Brunzelle JS, Houtz RL, Trievel RC. Structural origins for the product specificity of SET domain protein methyltransferases Proc Natl Acad Sci U S A 2008; 105: 20659-64.]. Interestingly, this switch was recently reported as a frequent somatic mutation in lymphoma, changing the EZH2 from a multifunctional mono- di- and trimethylase to an enzyne with increased trimethylase activity, but little or no mono- and dimethylase activity [35Yap DB, Chu J, Berg T, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation Blood 2011; 117(8
): 2451-9., 36Sneeringer CJ, Scott MP, Kuntz KW, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas Proc Natl Acad Sci U S A 2010; 107: 20980-5.]. Inhibitors specifically recognizing the mutant enzyme may be of interest.
 |
Fig. (7) Catalysis and control of methylation specificity. Backbone carbonyl oxygens and a catalytic tyrosine (orange) surrounding the departing methyl group of SAM (yellow) favor a nucleophilic attack by the ε-nitrogen of the substrate lysine (magenta), which must have been de-protonated beforehand [8Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications Biochim Biophys Acta 2009; 1789: 45-57.]. A limited number of residues (green, gray, cyan) restrict the alignment of a lone-pair on the accepting nitrogen with the scissile sulfur-methyl bond, a geometry necessary for methyl transfer to occur. Mutational analyses reported for G9a [18Wu H, Min J, Lunin VV, et al. Structural biology of human H3K9 methyltransferases PLoS One 2010; 5: e8570., 33Collins RE, Tachibana M, Tamaru H, et al. In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases J Biol Chem 2005; 280: 5563-70.], SETD7 [14Xiao B, Jing C, Wilson JR, et al. Structure and catalytic mechanism of the human histone methyltransferase SET7/9 Nature 2003; 421: 652-., 37Zhang X, Yang Z, Khan SI, et al. Structural basis for the product specificity of histone lysine methyltransferases Mol Cell 2003; 12: 177-85.], SETD8 [34Couture JF, Dirk LM, Brunzelle JS, Houtz RL, Trievel RC. Structural origins for the product specificity of SET domain protein methyltransferases Proc Natl Acad Sci U S A 2008; 105: 20659-64.], MLL [21Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks Mol Cell 2009; 33: 181-91.] and somatic mutations reported in EZH2 [35Yap DB, Chu J, Berg T, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation Blood 2011; 117(8 ): 2451-9.] confirm that these residues control the final methylation product: mono-, di- or tri-methyl lysine (Kme, Kme2, or Kme3 respectively). |
DRUG DESIGN
The cofactor and substrate peptide bind at two distinct pockets and meet at the catalytic site (Fig. 2, top). This suggests two avenues for drug design: competitive inhibition of cofactor or peptide binding. Potent small molecule inhibitors can only be developed if a site is druggable. Selective inhibition relies on the site’s diversity. Currently the very homologous enzymes GLP and G9a are the only two lysine PMTs that were crystallized in complex with substrate peptide competitors: Bix-01294 (IC50 ~1 µM), E67 (IC50 ~ 270 nM), E72 (IC50 ~ 100 nM), UNC0224 (IC50 ~ 15 nM) and UNC0638 (IC50 ~ 10 nM) (Fig. 1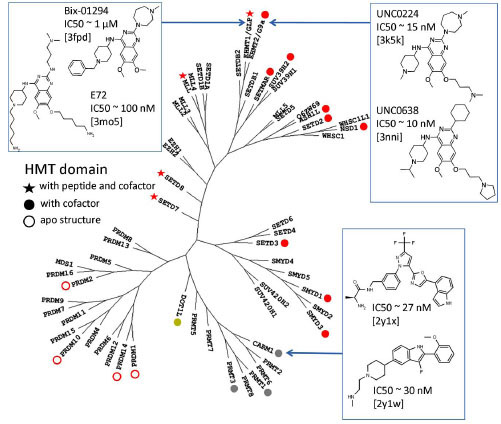 ) [25Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of
histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3., 26Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7., 38Chang Y, Ganesh T, Horton JR, et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases J Mol Biol 2010; 400: 1-7.-40Vedadi M, Barsyte-Lovejoy D, Liu F, et al. UNC0638: Potent,
selective, and cell-penetrant chemical probe of protein lysine
methyltransferases G9a and GLP Nat Chem Biol 2011.]. We used the program SiteMap (Schrodinger, NY) to evaluate the druggability of the pockets exploited by these inhibitors (Fig. 8
) [25Liu F, Chen X, Allali-Hassani A, et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of
histone lysine methyltransferase G9a J Med Chem 2009; 52: 7950-3., 26Chang Y, Zhang X, Horton JR, et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294 Nat Struct Mol Biol 2009; 16: 312-7., 38Chang Y, Ganesh T, Horton JR, et al. Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases J Mol Biol 2010; 400: 1-7.-40Vedadi M, Barsyte-Lovejoy D, Liu F, et al. UNC0638: Potent,
selective, and cell-penetrant chemical probe of protein lysine
methyltransferases G9a and GLP Nat Chem Biol 2011.]. We used the program SiteMap (Schrodinger, NY) to evaluate the druggability of the pockets exploited by these inhibitors (Fig. 8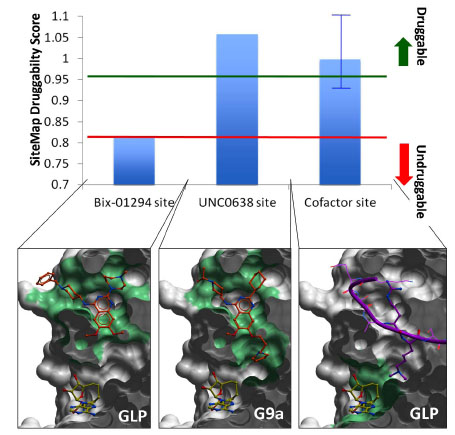 ). A druggability score (Dscore), validated against a large training set, is calculated as a function of volume, hydrophobicity, and enclosure of the site [41Halgren TA. Identifying and characterizing binding sites and assessing druggability J Chem Inf Model 2009; 49(2
): 377-89.]. A score larger than 0.95 indicates that the site is druggable; a value below 0.8 reflects poor druggability; a Dscore between 0.8 and 0.95 is in the gray zone, where no reliable conclusion can be drawn [41Halgren TA. Identifying and characterizing binding sites and assessing druggability J Chem Inf Model 2009; 49(2
): 377-89.]. Bix-01294 occupies the open section of the peptide binding groove, but does not exploit the lysine channel (Fig. 8). The druggability of the corresponding pseudo-site, which artificially excludes the lysine channel, is unclear. UNC0638, another peptide competitor, recapitulates the binding pose of Bix-01294, but has an additional aliphatic chain ending with a pyrrolidine that extends into the lysine channel (Fig. 8). With a Dscore of 1.05, the corresponding site is clearly druggable, as confirmed by the high potency of the ligand.
). A druggability score (Dscore), validated against a large training set, is calculated as a function of volume, hydrophobicity, and enclosure of the site [41Halgren TA. Identifying and characterizing binding sites and assessing druggability J Chem Inf Model 2009; 49(2
): 377-89.]. A score larger than 0.95 indicates that the site is druggable; a value below 0.8 reflects poor druggability; a Dscore between 0.8 and 0.95 is in the gray zone, where no reliable conclusion can be drawn [41Halgren TA. Identifying and characterizing binding sites and assessing druggability J Chem Inf Model 2009; 49(2
): 377-89.]. Bix-01294 occupies the open section of the peptide binding groove, but does not exploit the lysine channel (Fig. 8). The druggability of the corresponding pseudo-site, which artificially excludes the lysine channel, is unclear. UNC0638, another peptide competitor, recapitulates the binding pose of Bix-01294, but has an additional aliphatic chain ending with a pyrrolidine that extends into the lysine channel (Fig. 8). With a Dscore of 1.05, the corresponding site is clearly druggable, as confirmed by the high potency of the ligand.

 |
Fig. (8) Druggability of the peptide and cofactor binding sites. A druggability score was calculated with SiteMap (Schrodinger, NY) for the binding sites of the GLP/G9a inhibitors Bix-01294, UNC0638, and the cofactor (top) [41Halgren TA. Identifying and characterizing binding sites and assessing druggability J Chem Inf Model 2009; 49(2 ): 377-89.]. The peptide-binding site (center) is druggable, and can accommodate highly potent compounds, such as UNC0638. When the lysine channel is ignored, the druggability index drops sharply (left). The cofactor site (right) is also predicted to be druggable (blue bar represents the mean value across all available co-crystallized human SET domain PMT structures), but with varying druggability index from one enzyme to another (blue error bar), which reflects a variation in hydrophilicity and enclosure of the site. Green: pocket used to calculate the druggability score. The molecular surface of the enzyme is clipped across the Z-axis for better visualization. |
We also calculated the druggability of the cofactor binding site, defined as the pocket occupied by SAM, SAH, or the close analogue synefungin. The Dscore varied from 0.92 to 1.1 across all co-crystallized structures of human lysine PMTs, with a mean value of 1.0 (Fig. 8). While this site appears more druggable in some lysine PMTs than others, it is predicted to be druggable in all cases. The SETMAR structure is an exception, with a Dscore of 0.92, due to its particularly high hydrophilicity. This highlights a challenging feature shared by all enzymes. The cofactor site includes numerous polar groups that cannot be buried by hydrophobic ligands without significant desolvation penalty. These must be matched by a complex and specific network of hydrogen bond donors and acceptors decorating the inhibitor.
We have seen that in all available ternary structures, an arginine side-chain flanking the substrate lysine is an important contributor to binding enthalpy. It is interesting to note that the co-crystallized inhibitors all occupy the arginine binding site (Fig. 8), a feature that could inspire by analogy the design of SETD7 or SETD8 inhibitors (Fig. 4 bottom). Interaction hot spots that should be exploited by potent chemical inhibitors can be predicted based on receptor-ligand contacts conserved across all available structures. At the peptide binding site, a conserved double hydrogen-bond between the backbone of the substrate lysine and a beta-strand of the I-SET domain seems to be important for the interaction (Fig. 9A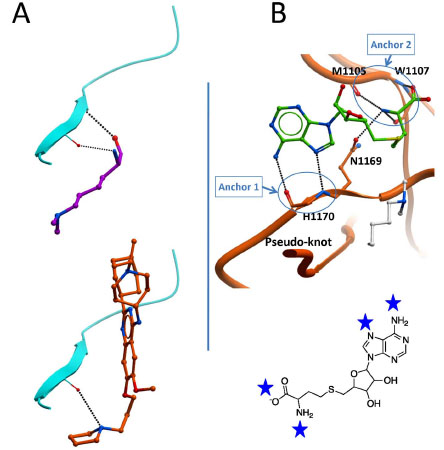 , top). Interestingly, this interaction is partially recapitulated by the pyrrolidine group of the potent inhibitor UNC0638 (Fig. 9A, bottom). At the cofactor binding site, a series of 6 hydrogen bonds engaged with five backbone atoms and one conserved asparagine side-chain of the SET domain is observed in all available structures (Fig. 9B, top). These hydrogen-bonds are clustered at two specific locations, acting as anchoring point for the cofactor, one at the adenine ring, the other at the methionine end. It is likely that potent inhibitors will need to mimic this profile of interaction.
, top). Interestingly, this interaction is partially recapitulated by the pyrrolidine group of the potent inhibitor UNC0638 (Fig. 9A, bottom). At the cofactor binding site, a series of 6 hydrogen bonds engaged with five backbone atoms and one conserved asparagine side-chain of the SET domain is observed in all available structures (Fig. 9B, top). These hydrogen-bonds are clustered at two specific locations, acting as anchoring point for the cofactor, one at the adenine ring, the other at the methionine end. It is likely that potent inhibitors will need to mimic this profile of interaction.

Selective inhibition can only be achieved if the structural chemistry of the pocket is sufficiently specific to a given enzyme. The peptide binding sites of lysine PMTs have evolved to recognize specific sequences. It is therefore reasonable to infer that structural features used to read specific sequences can be exploited to design selective inhibitors. This is in part confirmed by the selectivity profile of UNC0638 an inhibitor that specifically inhibit the H3K9 PMTs G9a and GLP, but not the H3K4 PMT SETD7, the H4K20 PMT SETD8, or even the H3K9 PMT SUV39H2 [40Vedadi M, Barsyte-Lovejoy D, Liu F, et al. UNC0638: Potent, selective, and cell-penetrant chemical probe of protein lysine methyltransferases G9a and GLP Nat Chem Biol 2011.]). The question of selectivity is not as clear for the cofactor site as it recognizes the same cofactor across all enzymes. The chemogenomic profiling of human kinases has demonstrated that selectivity can be engineered into ATP competitors. A recent study shows that the structural diversity of the SAM site in PMTs is comparable to that of the ATP site in kinases, suggesting that selective inhibition could be achieved at the PMT cofactor site [42Campagna-Slater V, Mok MW, Nguyen KT, Feher M, Najmanovich R, Schapira M. Structural chemistry of the histone methyltransferases cofactor binding site J Chem Inf Model 2011; 51(3 ): 612-23.]. The selectivity profile of chaetocin, a fungal metabolite that competes with SAM with some specificity for H3K9 PMTs, reinforces the hypothesis that selective inhibition at the cofactor site is chemically tractable [43Greiner D, Bonaldi T, Eskeland R, Roemer E, Imhof A. Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3-9 Nat Chem Biol 2005; 1: 143-5.].
Finally, the peptide and cofactor pockets could be simultaneously targeted by bi-substrate competitors, a mode ofaction that was proposed for existing non-SET domain PMT inhibitors [44Dowden J, Hong W, Parry RV, Pike RA, Ward SG. Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases Bioorg Med Chem Lett 2010; 20: 2103-5., 45Spannhoff A, Machmur R, Heinke R, et al. A novel arginine methyltransferase inhibitor with cellular activity Bioorg Med Chem Lett 2007; 17: 4150-3.].
CONCLUSION
We have highlighted general concepts regarding the structural mechanism of SET domain PMTs. A variety of domains can dress the core SET structure, and act as docking platforms for specific binding partners associated with diverse cellular events. Sequence and post-translational modification status of substrate peptides are mainly recognized by the I-SET domain, while a limited set of polar groups surrounding the substrate lysine control methylation specificity. The peptide and cofactor binding sites are chemically tractable, and can be targeted by selective small molecule inhibitors, independently or simultaneously. Conserved interaction patterns observed in co-crystal structures strongly suggest the presence of a discrete number of interaction hotspots that can be exploited to achieve potent inhibition.
FUNDING
The SGC is a registered charity (number 1097737) that receives funds from the Canadian Institutes for Health Research, the Canadian Foundation for Innovation, Genome Canada through the Ontario Genomics Institute, GlaxoSmithKline, Karolinska Institutet, the Knut and Alice Wallenberg Foundation, the Ontario Innovation Trust, the Ontario Ministry for Research and Innovation, Merck & Co., Inc., the Novartis Research Foundation, the Swedish Agency for Innovation Systems, the Swedish Foundation for Strategic Research and the Wellcome Trust.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGEMENTS
I would like to thank Cheryl Arrowsmith for comments on the manuscript, as well as Kong Nguyen and Calvin Santiago for computing druggability indices.