- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search




Current Chemical Genomics and Translational Medicine
(Discontinued)
ISSN: 2213-9885 ― Volume 12, 2018
Development of a Dehalogenase-Based Protein Fusion Tag Capable of Rapid, Selective and Covalent Attachment to Customizable Ligands
Lance P Encell*, #, 1, Rachel Friedman Ohana#, 1, Kris Zimmerman1, Paul Otto1, Gediminas Vidugiris1, Monika G Wood1, Georgyi V Los1, 3, Mark G McDougall2, Chad Zimprich1, Natasha Karassina1, Randall D Learish1, 4, Robin Hurst1, James Hartnett1, Sarah Wheeler1, Pete Stecha1, Jami English1, Kate Zhao1, 5, Jacqui Mendez1, Hélène A Benink1, Nancy Murphy1, Danette L Daniels1, Michael R Slater1, Marjeta Urh1, Aldis Darzins1, 6, Dieter H Klaubert2, Robert F Bulleit1, Keith V Wood1
Abstract
Our fundamental understanding of proteins and their biological significance has been enhanced by genetic fusion tags, as they provide a convenient method for introducing unique properties to proteins so that they can be examinedin isolation. Commonly used tags satisfy many of the requirements for applications relating to the detection and isolation of proteins from complex samples. However, their utility at low concentration becomes compromised if the binding affinity for a detection or capture reagent is not adequate to produce a stable interaction. Here, we describe HaloTag® (HT7), a genetic fusion tag based on a modified haloalkane dehalogenase designed and engineered to overcome the limitation of affinity tags by forming a high affinity, covalent attachment to a binding ligand. HT7 and its ligand have additional desirable features. The tag is relatively small, monomeric, and structurally compatible with fusion partners, while the ligand is specific, chemically simple, and amenable to modular synthetic design. Taken together, the design features and molecular evolution of HT7 have resulted in a superior alternative to common tags for the overexpression, detection, and isolation of target proteins.
Article Information
Identifiers and Pagination:
Year: 2012Volume: 6
Issue: Suppl 1
First Page: 55
Last Page: 71
Publisher Id: CCGTM-6-55
DOI: 10.2174/1875397301206010055
Article History:
Received Date: 02/3/2012Revision Received Date: 04/4/2012
Acceptance Date: 16/4/2012
Electronic publication date: 5/10/2012
Collection year: 2012
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Promega Corporation, 2800 Woods Hollow Road, Madison, WI 53711, USA; Tel: 608-274-1181; Fax: 608-298-4818; E-mail: lance.encell@promega.com# These authors contributed equally to this work.
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 02-3-2012 |
Original Manuscript | Development of a Dehalogenase-Based Protein Fusion Tag Capable of Rapid, Selective and Covalent Attachment to Customizable Ligands | |
INTRODUCTION
Proteins are critical for nearly all biological processes, yet for many we lack a solid understanding of their significance inside living cells. To elucidate function we need tools for studying proteins under different physiological conditions. It is essential to be able to purify proteins of interest as well as visualize their intracellular localization, dynamics, and interactions. Purification and visualization are challenging because it is difficult to distinguish individual proteins from the myriad of other proteins and biomolecules inside cells. A common solution is to append genetic fusion tags to proteins of interest so they can be examined in isolation. Fluorescent proteins have been widely used for this purpose [1 Tsien RY. The green fluorescent protein Annu Rev Biochem 1998; 67: 509-44.], as have a variety of other tags (e.g. FLAG, c-myc, poly-His, GST and MBP) that provide a means to label or capture proteins [2 Hammarstrom M, Hellgren N, van Den Berg S, Berglund H, Hard T. Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli Protein Sci 2002; 11(2 ): 313-21.-5 Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems Appl Microbiol Biotechnol 2003; 60(5 ): 523-33.].
The binding efficiency between commonly used fusion tags and their ligands is sufficient when the tag is highly abundant, but tagged proteins are frequently present at relatively low concentrations in biological samples. For example, recombinant genes are generally expressed poorly in cultured mammalian cells. Although E. coli can improve expression levels [6 Janssen DB, Scheper A, Dijkhuizen L, Witholt B. Degradation of halogenated aliphatic compounds by Xanthobacter autotrophicus GJ10 Appl Environ Microbiol 1985; 49(3 ): 673-7.], it lacks the machinery for introducing post-translational modifications necessary for proper folding of many eukaryotic proteins, often resulting in insoluble, unstable, or non-functional protein [7 Schindler JF, Naranjo PA, Honaberger DA, et al. Haloalkane dehalogenases: steady-state kinetics and halide inhibition Biochemistry 1999; 38(18 ): 5772-8.]. In these common situations where tagged target protein is not highly abundant, the utility of affinity tags can be limited by their binding affinity, selectivity, and kinetics [8 Kulakova AN, Stafford TM, Larkin MJ, Kulakov LA. Plasmid pRTL1 controlling 1-chloroalkane degradation by Rhodococcus rhodochrous NCIMB13064 Plasmid 1995; 33(3 ): 208-17.]. These limitations are inherent to the equilibrium-based nature of the binding between affinity tags and their binding ligands. Because these interactions are reversible, a portion of any tagged protein of interest will always remain unbound. The removal of this unbound portion (e.g. during washes) further exacerbates the situation, as it causes additional tagged protein to become unbound as the sample re-equilibrates.
Binding would be more efficient if the reaction between tag and ligand was rapid, selective, and irreversible. The high affinity interaction between streptavidin and biotin exemplifies these desirable characteristics. However, streptavidin is limited as a fusion tag because of its tetrameric structure. When genetically appended onto another protein, the resulting monomeric form loses much of its binding affinity [9 Kulakova AN, Larkin MJ, Kulakov LA. The plasmid-located haloalkane dehalogenase gene from Rhodococcus rhodochrous NCIMB 13064 Microbiology 1997; 143(Pt 1): 109-5.]. To improve upon current tags, we adopted a protein design concept based on hydrolytic enzymes to enable rapid and irreversible attachment to a unique synthetic ligand. Hydrolases catalyze nucleophilic displacements to produce covalent enzyme-substrate intermediates. These intermediates are resolved by an activated water molecule to yield the reaction products. Altering the amino acids required for water activation can block hydrolysis and product release, and in doing so result in a stable, covalent protein adduct. Because a substrate cannot be turned over it becomes a ligand capable of binding to or capturing the mutant hydrolase. We focused on haloalkane dehalogenases, enzymes that catalyze the breakdown of haloalkanes [6 Janssen DB, Scheper A, Dijkhuizen L, Witholt B. Degradation of halogenated aliphatic compounds by Xanthobacter autotrophicus GJ10 Appl Environ Microbiol 1985; 49(3 ): 673-7., 7 Schindler JF, Naranjo PA, Honaberger DA, et al. Haloalkane dehalogenases: steady-state kinetics and halide inhibition Biochemistry 1999; 38(18 ): 5772-8.]. These enzymes are small, monomeric, and not found in eukaryotic systems [8 Kulakova AN, Stafford TM, Larkin MJ, Kulakov LA. Plasmid pRTL1 controlling 1-chloroalkane degradation by Rhodococcus rhodochrous NCIMB13064 Plasmid 1995; 33(3 ): 208-17.-11 Gray KA, Richardson TH, Robertson DE, Swanson PE, Subramanian MV. Soil-based gene discovery: a new technology to accelerate and broaden biocatalytic applications Adv Appl Microbiol 2003; 52: 1-27.]. Moreover, their substrates should be effective ligands. Because they are chemically simple, straightforward synthetic methods can be used to attach different functionalities. This makes them well suited to become modular binding partners. These substrates are also generally membrane permeant, making them suitable for use with live cells. In considering different dehalogenases we chose the enzyme from Rhodococcus (DhaA) because it is known to have broad substrate specificity [7 Schindler JF, Naranjo PA, Honaberger DA, et al. Haloalkane dehalogenases: steady-state kinetics and halide inhibition Biochemistry 1999; 38(18 ): 5772-8., 12 Yokota T, Omori T, Kodama T. Purification and properties of haloalkane dehalogenase from Corynebacterium sp. strain m15-3 J Bacteriol 1987; 169(9 ): 4049-54., 13 Bosma T, Kruizinga E, de Bruin EJ, Poelarends GJ, Janssen DB. Utilization of trihalogenated propanes by Agrobacterium radiobacter AD1 through heterologous expression of the haloalkane dehalogenase from Rhodococcus sp. strain M15-3 Appl Environ Microbiol 1999; 65(10 ): 4575-81.]. The promiscuous nature of DhaA suggests it could potentially react with haloalkanes appended with modular functionalities.
DhaA carries out dehalogenation using a serine protease-like catalytic triad [14 Newman J, Peat TS, Richard R, et al. Haloalkane dehalogenases:
structure of a Rhodococcus enzyme Biochemistry 1999; 38(49
): 16105-4.-16 Janssen DB. Evolving haloalkane dehalogenases Curr Opin Chem
Biol 2004; 8(2
): 150-9.]. Initially, a nucleophilic Asp attacks the α -carbon of the substrate (Fig. 1A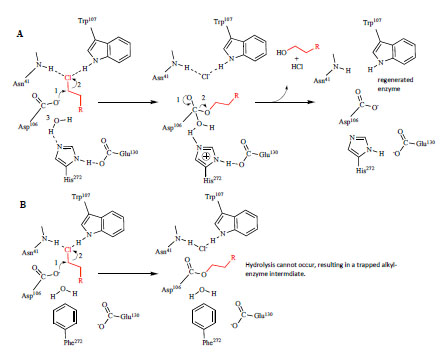 ), producing a covalent alkyl-enzyme ester intermediate. A nearby His, acting as a general base, catalyzes hydrolysis of this intermediate. Depending on the species, Asp or Glu completes the triad, providing structure as well as stabilization to the positive charge formed on the His ring. In the final (and commonly rate-limiting) step of the reaction, products (i.e. halide and R-OH) are released from the active site, resulting in enzyme regeneration [17 Bosma T, Pikkemaat MG, Kingma J, Dijk J, Janssen DB. Steadystate
and pre-steady-state kinetic analysis of halopropane conversion
by a rhodococcus haloalkane dehalogenase Biochemistry
2003; 42(26
): 8047-53., 18 Lau EY, Kahn K, Bash PA, Bruice TC. The importance of reactant
positioning in enzyme catalysis: a hybrid quantum mechanics/
molecular mechanics study of a haloalkane dehalogenase Proc
Natl Acad Sci USA 2000; 97(18
): 9937-42.]. It was previously shown with the dehalogenase from Xanthobacter, DhlA, that mutating the catalytic His yields a variant that forms a stable ester bond with 1,2-dibromoethane [19 Pieters RJ, Fennema M, Kellogg RM, Janssen DB. Design and
synthesis of reagents for phage display screening of dehalogenases
Bioorg Med Chem Lett 1999; 9(2
): 161-6., 20 Pries F, Kingma J, Krooshof GH, Jeronimus-Stratingh CM, Bruins
AP, Janssen DB. Histidine 289 is essential for hydrolysis of the alkyl-
enzyme intermediate of haloalkane dehalogenase J Biol Chem
1995; 270(18
): 10405-1.]. We replaced the analogous His in DhaA with Phe (Fig. 1B), and the resulting variant formed a similar covalent attachment to haloalkanes [21 Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein
labeling technology for cell imaging and protein analysis ACS
Chem Biol 2008; 3(6
): 373-82.].
), producing a covalent alkyl-enzyme ester intermediate. A nearby His, acting as a general base, catalyzes hydrolysis of this intermediate. Depending on the species, Asp or Glu completes the triad, providing structure as well as stabilization to the positive charge formed on the His ring. In the final (and commonly rate-limiting) step of the reaction, products (i.e. halide and R-OH) are released from the active site, resulting in enzyme regeneration [17 Bosma T, Pikkemaat MG, Kingma J, Dijk J, Janssen DB. Steadystate
and pre-steady-state kinetic analysis of halopropane conversion
by a rhodococcus haloalkane dehalogenase Biochemistry
2003; 42(26
): 8047-53., 18 Lau EY, Kahn K, Bash PA, Bruice TC. The importance of reactant
positioning in enzyme catalysis: a hybrid quantum mechanics/
molecular mechanics study of a haloalkane dehalogenase Proc
Natl Acad Sci USA 2000; 97(18
): 9937-42.]. It was previously shown with the dehalogenase from Xanthobacter, DhlA, that mutating the catalytic His yields a variant that forms a stable ester bond with 1,2-dibromoethane [19 Pieters RJ, Fennema M, Kellogg RM, Janssen DB. Design and
synthesis of reagents for phage display screening of dehalogenases
Bioorg Med Chem Lett 1999; 9(2
): 161-6., 20 Pries F, Kingma J, Krooshof GH, Jeronimus-Stratingh CM, Bruins
AP, Janssen DB. Histidine 289 is essential for hydrolysis of the alkyl-
enzyme intermediate of haloalkane dehalogenase J Biol Chem
1995; 270(18
): 10405-1.]. We replaced the analogous His in DhaA with Phe (Fig. 1B), and the resulting variant formed a similar covalent attachment to haloalkanes [21 Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein
labeling technology for cell imaging and protein analysis ACS
Chem Biol 2008; 3(6
): 373-82.].
 |
Fig. (1) Catalytic mechanism of dehalogenation by DhaA and strategy for trapping the covalent intermediate. A. In the first step of catalysis, the nucleophile, Asp106 attacks the α-carbon of the chloroalkane (shown in red) to produce a covalent, alkyl-enzyme intermediate. His272 catalyzes hydrolysis of the intermediate, the release of products from the active site, and regeneration of enzyme. Glu130 provides structure at the active site and stabilizes the positive charge on His272 that forms during hydrolysis. In addition to forming the halide binding site, Trp107 and Asn41 stabilize the Cl- leaving group following hydrolysis [16 Janssen DB. Evolving haloalkane dehalogenases Curr Opin Chem Biol 2004; 8(2 ): 150-9.]. B. The strategy for trapping the covalent intermediate was to replace His272 with a residue (e.g. Phe) that cannot act as a general base, and therefore cannot hydrolyze the alkyl-enzyme intermediate. |
Configuring the mutant dehalogenase into a useful fusion tag required optimization of the ligand as well as the protein. We used a computational model of DhaA for designing a suitable ligand and also for guiding mutagenesis at the protein’s binding tunnel that resulted in rapid and efficient binding to a ligand containing either a fluorophore or a biotin solid support. Because it was critical for the tag to be compatible with different fusion partners, we used additional molecular evolution to optimize the structure of the tag and define peptide linkers that could be used to fuse the tag to either terminus of a target protein. In addition to helping maintain the structural and functional integrity of both the target protein and the tag, the linker was further designed to contain an optimized proteolytic cleavage site to enable downstrean removal of the tag.
The resulting variant and linker, generally referred to as HaloTag® (HT7), is a robust genetic fusion tag utilizing an irreversible attachment to a ligand to provide highly efficient binding, and as a result overcomes the limitations associated with common, equilibrium-based affinity tags. In addition, HT7 provides technical features that impart reliable performance under varied experimental circumstances. It has broad structural compatibility with fused partner proteins and binds with high selectivity to its cognate ligand. HT7 is small, monomeric, and mechanistically orthogonal to the experimental host, making it generally inert to relevant biological systems of study. Furthermore its binding ligand is chemically simple and capable of carrying diverse functionalities, enabling both protein capture and visualization. HT7 should have broad applicability in areas related to the biochemical characterization of recombinant proteins as well as the detection and analysis of proteins in live cells or animals.
MATERIALS AND METHODOLOGIES
Bacterial Strains, Genetic Materials, and Reagents
E. coli strains JM109 and KRX ([F´, traD36, ΔompP, proA+B+, lacIq , Δ(lacZ)M15] ΔompT, endA1, recA1, gyrA96 (Nalr), thi-1, hsdR17 (rk–, mk+), e14– (McrA–), relA1, supE44, Δ(lac-proAB), Δ(rhaBAD)::T7 RNA polymerase) were from Promega. All chemicals were from Sigma-Aldrich unless otherwise noted. Enzymes and other reagents were from Promega unless otherwise noted. RhodococcusdhaA (in pET-3a) was a generous gift from Dr. Clifford J. Unkefer (Los Alamos National Laboratory). Dulbecco’s modified essential medium (DMEM), F12, and fetal bovine serum (FBS) were from Life Technologies. 24-well plates were from Nalge Nunc International. LT1 transfection reagent was from Mirus Bio. Protein molecular weight markers were from Pierce. Mammalian cell lines were from ATCC.
Chloroalkane Substrates and Ligands
Synthesis of FAM-14-Cl (FAM-ligand) and TMR-14-Cl (TMR-ligand) (Fig. 2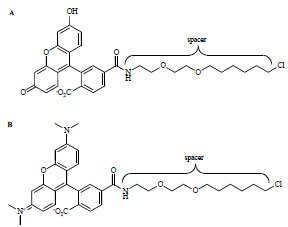 ) was previously described [21 Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein
labeling technology for cell imaging and protein analysis ACS
Chem Biol 2008; 3(6
): 373-82.]. The TMR-ligand and the Oregon Green-ligand are available from Promega. Synthesis of the PEG Biotin-ligand was previously described [22 Svendsen S, Zimprich C, McDougall MG, Klaubert DH, Los GV.
Spatial separation and bidirectional trafficking of proteins using a
multi-functional reporter BMC Cell Biol 2008; 9: 17.] and this ligand is also available from Promega. The preparations of other chloroalkanes are described in the
Supplementary Material.
) was previously described [21 Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein
labeling technology for cell imaging and protein analysis ACS
Chem Biol 2008; 3(6
): 373-82.]. The TMR-ligand and the Oregon Green-ligand are available from Promega. Synthesis of the PEG Biotin-ligand was previously described [22 Svendsen S, Zimprich C, McDougall MG, Klaubert DH, Los GV.
Spatial separation and bidirectional trafficking of proteins using a
multi-functional reporter BMC Cell Biol 2008; 9: 17.] and this ligand is also available from Promega. The preparations of other chloroalkanes are described in the
Supplementary Material.
 |
Fig. (2) Substrate/ligand chemical structures. A. FAM-14-Cl (FAM-ligand; Eex/Eem=490/520 nm). B. TMR-14-Cl (TMR-ligand; Eex/Eem=545/575 nm). |
dhaA Cloning and Vectors (see Supplementary Material) Site-Directed Mutagenesis
Mutagenesis was carried out using QuickChange (Agilent). Oligonucleotides were from Integrated DNA Technologies. Oligonucleotides containing NNK or RVN codons (N=A, T, C or G; K=G or T; R=A or G; V=A, G, or C) were designed and synthesized to saturate a parental sequence codon of interest with multiple amino acids. Mutagenesis reactions were used to transform E. coli JM109 or KRX, and then plasmid DNA was isolated and sequenced. An ample number of colonies were sequenced to verify library quality and demonstrate non-biased distribution of substitutions for a particular codon. Combined sequences were constructed by either transferring the relevant mutations from one plasmid to another using restriction enzyme digestion, agarose gel purification, and ligation, or by QuickChange Multi (Agilent).
Bacterial Expression and Lysate Preparation
Variants in pGEX-5X3 (GE Healthcare) were overexpressed in E. coli JM109 at 25 oC according to the vector manufacturer’s protocol. Cells were harvested and stored at -70 oC. Variants in pF-based vectors (in E. coli KRX) were grown overnight at 30 oC in 2 ml LB + kanamycin (25 µg/ml) or ampicillin (100 µg/ml) and diluted back 1:100 to fresh media and grown at 37 oC to an OD600 of 0.5. Rhamnose was added to a final concentration of 0.2% (w/v) and the cells induced for variable times at either 25 or 30 oC.
GST-based Affinity Purification (see Supplementary Material) DhaA Activity Assay
For optimization of the spacer component of the substrates and ligands, DhaA hydrolase activity was measured using a halide release assay previously described [23 Holloway P, Trevors JT, Lee H. A colorimetric assay for detecting haloalkane dehalogenase activity J Microbiol Methods 1998; 32(1 ): 31-6.]. Following the addition of affinity purified DhaA to a cocktail containing substrate and phenol red indicator, halide release in the form of HCl was monitored colorimetrically at 558 nm and initial rates of acid production were calculated based on a standard curve for HCl.
Protein Labeling and Analysis
Purified proteins, bacterial lysates, or cultured mammalian cells were incubated with the TMR-ligand for various lengths of time at 25 oC. Reactions were stopped by adding SDS gel loading buffer (final [SDS]=0.5%, w/v). Following a 2 min exposure to 95 oC, aliquots were resolved by SDS-PAGE (4–20% Tris-glycine (BioRad)) and scanned (Eex/Eem=532/580 nm) for labeling (i.e. functional expression) using a Typhoon fluorescence scanner (GE Healthcare). Quantitation of fluorescence bands was performed using ImageQuant software (GE Healthcare). SimplyBlue SafeStain (Life Technologies) was used for total protein imaging (Eex=633 nm, no emission filter) using the same scanner.
Mass Spectrometry Analysis of TMR-Labeled Protein
MW determination of proteins was performed at the Mass Spectrometry Facility (Biotechnology Center, University of Wisconsin-Madison). The methodologies and details for these analyses can be found in the Supplementary Material. Mass analysis for the ribosome pull-down experiments was carried out by NextGen Sciences.
HPLC Gel Permeation Analysis of Protein Aggregation
A comparison of the relative hydrodynamic volumes of non-fused HT2 and HT7 to that of three protein standards (BSA, 66 kDa; ovalbumin, 43 kDa; and bovine pancreas ribonuclease, 14 kDa) was made by gel permeation chromatography on a Hewlett Packard 1050 HPLC using a 4.6 x 250 mm Macrosphere GCP 100A column (Alltech) and a mobile phase of PBS (flow rate of 0.25 ml/min). Protein was monitored at 214 and 280 nm.
Kinetic Analysis of Protein-Ligand binding by Fluorescence Polarization (FP)
Purified variant protein (excess) was incubated with ligand (TMR-14-Cl/TMR-ligand or FAM-14-Cl/FAM-ligand) in PBS + 0.01% CHAPS, and binding was monitored over time by FP (TMR: Eex/Eem=535/580 nm; FAM: Eex/Eem=485/535 nm) using an Ultra plate reader (Tecan). Apparent rate constants were calculated from the second order rate equation,
kt=(1/B0-A0)ln(B0-x)A0/(A0-x)B0
Computational Structure Models
Molecular modeling was performed using InsightII 2000 software (Accelrys Software Inc.). Homology models were built with Modeler using the x-ray crystallographic structure of DhaA (PDB code 1BN6) as a template. Substrates/ligands were manually docked into the models and covalently bonded to the carboxylate oxygen of the Asp106 nucleophile. The models were energy minimized with Discover-3 (CFF91 force field) using non-bond interactions with group-based or atom-based cutoffs, a distance-dependent dielectric of 1.0, and a final convergence of 0.01. During energy minimization, protein residues at a distance greater than 8 Å from the ligand were fixed, and harmonic Cα restraints were applied to the remaining residues. For all minimized models, bump checks (atom overlaps greater than 10% of atom van der Waals radii) were performed between the ligand and residues within 8 Å of the ligand to determine steric hindrances. The substrate/ligand entry tunnel was visualized by calculating a Connolly surface with a probe radius of 1.4 Å for residues within 5 Å of the substrate/ligand. Models were superimposed structurally to evaluate changes in the position of specific residues.
Screening for Mutants with Improved Ligand Binding Rates
Overnight cultures (LB, 30 oC, 96-well microtiter plates) of variants (pGEX-5X3) were diluted 1:40 into fresh terrific broth + ampicillin + 0.1 mM IPTG and grown overnight at 30 ° with shaking. The next day, cultures were harvested and supernatants removed. Cell pellets were resuspended in a cocktail of MagneGST (Promega) paramagnetic resin, FastBreak lysis reagent, and the TMR-ligand (15 µM). Resin binding capacity was intentionally limiting so that a fixed amount of each mutant was captured in each well. After a 10 min incubation with mild shaking, resin was washed three times with PBS + 0.1% Tween-20 (PBST) using the assistance of a magnet. Note that the reaction between excess TMR-ligand and unbound protein did not contribute to the final signal because the binding of protein to resin was so fast. The binding between H272F and ligand was linear for up to 30 min, indicating that the 10 min incubation time used for the screen was well within the linear range of the binding assay. Bound protein was eluted from the resin with glutathione-containing MagneGST elution reagent (Promega) by incubating for 5 min with mild shaking at 25 oC, and eluents were transferred to a new plate for fluorescence measurement (Eex/Eem=550/580 nm) using a Safire fluorescent plate reader (Tecan) configured within a Freedom robotic workstation (Tecan). Clones demonstrating at least 20% improvement in binding rate over the parental clone (H272F) were streaked to fresh agar and four random colonies for each hit were validated in a secondary screen using the same assay.
Luminescence Measurements
Humanized Renilla luciferase (RLuc) activity was measured using the Renilla Luciferase Assay System (Promega) according to the vendor protocols. Diluted bacterial lysates were assayed using injectors on a GloMax 96 Microplate Luminometer. Light emission was integrated over 10 sec after an initial 2 sec pre-read delay.
Random Mutagenesis
Random mutagenesis of the entire gene was carried out using error-prone PCR (GeneMorph II; Agilent). Libraries were generated to contain on average 2–3 mutations per kb. Additional details can be found in the Supplementary Material.
Screens for Improved Functional Expression
Libraries (pF1K+; see Supplementary Material) propagated in E. coli KRX were picked into 96-well microtiter plates and grown for 20 h at 30 oC in LB + antibiotic. The next day the cultures were used to inoculate (1:20) autoinduction media: M9 + glycerol (0.2%), gelatin (1%), rhamnose (0.2%), glucose (0.025%), and antibiotic. These cultures were grown at 25 oC for 22 h. Under these conditions, expression of the chromosomal copy of T7 RNA polymerase is repressed until glucose is consumed (12 h). At this time, rhamnose activates the expression of T7 RNA polymerase and expression begins (i.e. 10 h induction. Induced libraries were lysed for 30 min using a cocktail of MagneGST lysis buffer (0.5x), lysozyme (1 mg/ml), and RQ Dnase I (10 units), and then assayed for FAM-ligand (7.5 nM) binding at 7 min (linear range) using FP (Eex/Eem= 485/535 nm) on a Tecan GENios Pro reader, or the amount of total functional fusion protein based on TMR-labeling to completion (20 µM TMR-ligand, 1 h, 25 oC) and SDS-PAGE/fluorescence scanning. Secondary screens for validating hits were carried out using lysates from cells induced at more stringent temperatures (30 or 37 oC). Note that in the first round of mutagenesis on the HT2 template, variants were screened in the context of C-terminal chloramphenicol acetyltransferase. The intention was that this would offer a positive genetic selection for more stable, properly folded fusion protein [25 Sieber V. Selection for soluble proteins via fusion with chloramphenicol acetyltransferase Methods Mol Biol 2003; 230: 45-55.], however we were unable to find conditions to make the selection work in our system.
C-Terminus and Linker Optimization
Both the C-terminus and linker variants were created by either direct ligation of duplex oligonucleotides containing desired mutations or by random mutagenesis (see Supplementary Material).
TEV Protease Cleavage Assay
Soluble fractions of bacterial lysates were labeled to completion using 20 µM TMR-ligand and incubated with 0.5 units of ProTEV protease (Promega) for 30 min at 30 oC in a buffer containing 50 mM HEPES (pH 7), 1 mM DTT, and 0.5 mM EDTA. Cleavage efficiency was monitored by SDS-PAGE and fluorescence scanning.
Circular Dichroism (CD) Measurements
Purified HT2 or HT7 were dissolved in 50 mM sodium phosphate buffer (pH 7) at either 306 ng/µl (HT2) or 166 ng/µl (HT7); CD measurements used 0.1 cm cuvettes. An Aviv 202SF CD spectrophotometer equipped with a Peltier temperature controlled multicell rotor (Biophysics Instrumentation Facility, University of Wisconsin-Madison) was used to record spectra as a function of temperature. With both samples in the rotor, the temperature was increased from 8–83 oC in 3 oC steps (0.5 oC deadband; 2 min equilibration once in the deadband) and CD spectra recorded from 195–260 nm (2 nm steps with 3 sec averaging time).
Stability of HT2 and HT7
Purified HT2 or HT7 were exposed to elevated temperatures for 30 min and then immediately measured for remaining activity using the FP-based FAM-ligand binding assay. A pulse proteolysis method [26 Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding Nat Methods 2005; 2(3 ): 207-12.] was used to measure the stability of the proteins following exposure to urea. This approach, based on the sensitivity of unfolded protein to cleavage by thermolysin, was used to measure the ability of the proteins to retain proper folding upon exposure to these agents. TMR-labeled protein was exposed to varying concentrations of urea overnight at 25 oC and treated with the protease, thermolysin (2 min; quenched by EDTA). Samples were resolved by SDS-PAGE and analyzed by fluorescence scanning.
Isolation and Characterization of Ribosomes
Human RPS9 (NM_001013.2) was obtained from Genecopoeia. HEK-293T were used for ribosome isolation, HEK-293T cells stably expressing luciferase were used for ribosome isolation for translational studies, and U2OS cells stably expressing RPS9-HT7 were used for imaging. All were maintained in DMEM supplemented with 10% FBS at 37 °C in an atmosphere of 5% CO2. Cells were transfected using FuGENE HD transfection reagent (Promega) according to the manufacturer’s protocols. For isolating ribosomes, cells (1.2 x 107) were plated in a 15 cm plate. After reaching 70–80% confluency (18–24 h) cells were transfected with the RPS9-HT7 fusion construct (pFC14; Promega) or a HT7 control vector. 24 h post-transfection, cells were harvested and frozen at -80°C until processing. Pull-down experiments were performed according to the manufacturer’s guidelines (http://www.promega.com/tbs/tm342/tm342.pdf) with the exception of supplementing lysis, wash, and elution buffers with 30 mM MgCl2 and 40 units/ml of RNasin (Promega). Captured complexes were liberated by ProTEV (Promega) cleavage at the RPS9-HT7 linker and analyzed by SDS-PAGE/silver staining and LC/MS/MS (NextGen Sciences). For imaging experiments, U2OS cells stably expressed RPS9-HT7 were serum-starved (DMEM) for 18 h and labeled with 5 µM TMR-ligand in serum-free media for 15 min at 37 °C and 5% CO2. Cells were washed twice with pre-warmed, 37 °C complete media (DMEM + 10% FBS) to remove residual TMR-ligand and then given complete media and placed back at 37 °C and 5% CO2 for recovery. At either 3 or 24 h post-recovery, cells were treated with 5 µM Oregon Green-ligand in complete media for 15 min at 37 °C and 5% CO2 to label the new populations of RPS9-HT7. Cells were washed twice with pre-warmed complete media and imaged. Images were acquired on a Fluoview FV500 confocal microscope (Olympus) containing a 37 °C + CO2 environmental chamber (Solent Scientific) using appropriate filter sets. In vitro ribosomal translation assays were performed using PURExpressed Δ Ribosome kit (NEB) with the following modifications: (a) Fluc mRNA was added to the native ribosomes control, and (b) for RPS9-HT7, the HT7 control, and the untransfected controls, native ribosomes were excluded and substituted with RPSP9-HT7, HT7, or untransfected cells. In vitro translation reactions were carried out at 30 oC for 2 h and then assayed for luciferase activity (Luciferase assay reagent, Promega).
RESULTS
Ligand Design and Optimization
The crystal structure of DhaA [14 Newman J, Peat TS, Richard R, et al. Haloalkane dehalogenases: structure of a Rhodococcus enzyme Biochemistry 1999; 38(49 ): 16105-4.] indicates that the enzyme active site is buried deep within the enzyme, suggesting that a ligand designed for a non-catalytic variant of DhaA would require a spacer segment to prevent steric hindrance with attached synthetic functionalities. To examine this further we created a computational model of the enzyme’s substrate binding tunnel by superimposing multiple published structures of related dehalogenases complexed with different substrates containing 2–4 carbons [27 Verschueren KH, Kingma J, Rozeboom HJ, Kalk KH, Janssen DB, Dijkstra BW. Crystallographic and fluorescence studies of the interaction of haloalkane dehalogenase with halide ions Studies with halide compounds reveal a halide binding site in the active site Biochemistry 1993; 32(35 ): 9031-7.-30 Streltsov VA, Prokop Z, Damborsky J, Nagata Y, Oakley A, Wilce MC. Haloalkane dehalogenase LinB from Sphingomonas paucimobilis UT26: X-ray crystallographic studies of dehalogenation of brominated substrates Biochemistry 2003; 42(34 ): 10104-2.]. The collective positions of these substrates allowed us to infer that they likely enter through the helical cap domain. This access tunnel measured approximately 15 Å from the surface of the enzyme to the catalytic nucleophile, suggesting longer ligands (>4 carbons) would be required to bind the protein without interference from the functional group.
We tested a panel of chloroalkanes (Cl-(CH2)n) and chloroalcohols (Cl-(CH2)n-OH) to determine whether longer molecules could be substrates for DhaA (purified as a fusion to GST). Chloro compounds were chosen over other halides (e.g. bromo, iodo) because they are generally less reactive substrates [31 Schanstra JP, Kingma J, Janssen DB. Specificity and kinetics of
haloalkane dehalogenase J Biol Chem 1996; 271(25
): 14747-53.]. We observed that shorter chain compounds were better substrates for DhaA. However, there was measurable activity for both alkanes and alcohols containing as many as 10 carbons, demonstrating that both a chemical spacer and a polar functionality (-OH) were tolerated by the enzyme. We next synthesized a panel of chloroalkanes containing carboxyfluorescein (FAM) or carboxytetramethylrhodamine (TMR) fluorophores and spacers of different length and/or hydrophobicity. The most reactive substrates (FAM- and TMR-14-Cl; Fig (2)) contained a ~17 Å spacer consisting of two repeating polyethylene glycol moieties between the chlorine and the fluorophore.
Modifying DhaA to form a Stable Attachment with Chloroalkanes
To trap the covalent intermediate that forms between DhaA and chloroalkanes we replaced the catalytic base residue, His272, with four different amino acids predicted to prevent catalysis: Gln because it was previously shown to inactivate the related dehalogenase, DhlA [20 Pries F, Kingma J, Krooshof GH, Jeronimus-Stratingh CM, Bruins
AP, Janssen DB. Histidine 289 is essential for hydrolysis of the alkyl-
enzyme intermediate of haloalkane dehalogenase J Biol Chem
1995; 270(18
): 10405-1.], Phe because of its similar structure to His, and Ala and Gly because of their small size. We also changed the nucleophile (Asp106) to a cysteine so that a more stable thioether bond would form between the enzyme and substrate. Bacterial lysates containing the variants (fusions to GST) were incubated with a molar excess of FAM-14-Cl, resolved by SDS-PAGE, and analyzed for protein labeling using fluorescence scanning. Labeling was detected for each variant, suggesting each of the five was capable of forming an attachment to FAM-14-Cl (referred to from this point forward as the FAM-ligand) that was stable under the denaturing conditions (SDS, 95 °C) used for the gel analysis. Of the five variants tested the Phe272 mutant (H272F; Fig. 1B) reacted the most efficiently with the FAM-ligand.
Characterizing the Attachment between H272F and Chloroalkane Ligands
To determine if labeling was stoichiometric (i.e. one ligand per protein) we incubated H272F with a molar excess of FAM-ligand and characterized the products by mass spectrometry [32 Covey TR, Bonner RF, Shushan BI, Henion J. The determination
of protein, oligonucleotide and peptide molecular weights by ionspray
mass spectrometry Rapid Commun Mass Spectrom 1988; 2(11
): 249-56.]. The mass of the protein treated with ligand was 545 mass units higher than the untreated protein, a difference consistent with the expected mass gain predicted by the addition of a single FAM-ligand. Similar to the SDS-PAGE analysis, the processing associated with the mass analysis (e.g. organic solvents, acidic pH) provided evidence of a stable attachment between H272F and ligand. To investigate the specificity of ligand attachment we examined the binding reaction in a background of cellular proteins from both bacterial and mammalian cells. Bacterial lysates containing H272F (fusion to GST) were incubated with TMR-14-Cl (i.e. the TMR-ligand) and analyzed by SDS-PAGE and fluorescence scanning. We observed concentration (TMR-ligand) and time-dependent formation of a single predominant fluorescent product from the reaction (Figs. 3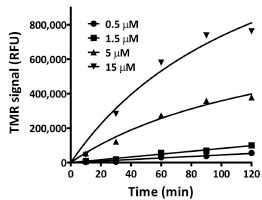 ,
S1A). In contrast, no products were detected from control lysates containing either the wild type DhaA enzyme or free GST tag. Binding was also specific in mammalian cells, as a TMR-labeled product could only be detected in CHO-K1 cells expressing H272F (Fig.
S1B).
,
S1A). In contrast, no products were detected from control lysates containing either the wild type DhaA enzyme or free GST tag. Binding was also specific in mammalian cells, as a TMR-labeled product could only be detected in CHO-K1 cells expressing H272F (Fig.
S1B).
 |
Fig. (3) Time and dose-dependent formation of a stable attachment between H272F and the TMR-ligand. Plot of fluorescence intensities (RFU) determined by SDS-PAGE and fluorescence scanning for TMR. The actual gel image can be found in the Supplementary Material (Fig. S1A). |
Improving Ligand Binding Efficiency
Despite the specificity and stability of the H272F-ligand attachment, our attempts to use this protein as an affinity handle for pull downs or as a tag for cellular imaging were unsuccessful. We considered the underlying cause of inefficient binding could be poor kinetics. To investigate this we measured the kinetics of the reaction between H272F (fusion to GST) and the TMR/FAM-ligands using fluorescence polarization (FP). Because FP measures the loss of free ligand from a sample, a significant molar excess of H272F (15 µM) over the ligands (15 nM) was necessary for these reactions. The TMR-ligand reaction required nearly 2 h to reach completion, while the FAM-ligand reaction required >10 h. The apparent second order rate constants for the TMR- and FAM-ligands were 67 and 3.0 M-1sec-1, respectively [33 Los GV, Wood K. The HaloTag: a novel technology for cell imaging and protein analysis Methods Mol Biol 2007; 356: 195-208.]. These values, which are >4 orders of magnitude lower than published values for streptavidin and biotin [34 Qureshi MH, Yeung JC, Wu S-C, Wong S-L. Development and characterization of a series of soluble tetrameric and monomeric streptavidin muteins with differential biotin binding affinities J Biol Chem 2001; 276(49 ): 46422-8.], provided a strong indication that more rapid binding was necessary for H272F to be a useful fusion tag.
To assist in optimizing H272F for faster ligand binding, we built a homology model of the protein based on the crystal structure of DhaA [14 Newman J, Peat TS, Richard R, et al. Haloalkane dehalogenases:
structure of a Rhodococcus enzyme Biochemistry 1999; 38(49
): 16105-4.]. A single TMR-ligand was manually docked into the model and a covalent bond created to the Asp106 nucleophile (Fig. 4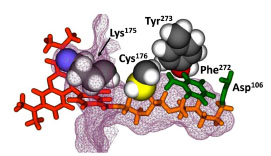 ). From a subset of amino acids within 5 Å of the bound ligand, Lys175, Cys176, and Tyr273 appeared to have the most contact with the ligand. It was reasonable to assume that decreasing the size of the side chain could open up the tunnel and facilitate more rapid ligand entry and binding. Additional evidence for the importance of positions 176 and 273 came from a previous report on the engineering of a DhaA variant containing C176Y and Y273F. This double mutant could bind more efficiently to 1,2,3-trichloropropane, suggesting a role for these residues in positioning incoming haloalkane substrates for efficient nucleophilic attack [35 Bosma T, Damborsky J, Stucki G, Janssen DB. Biodegradation of
1,2,3-trichloropropane through directed evolution and heterologous
expression of a haloalkane dehalogenase gene Appl Environ Microbiol
2002; 68(7
): 3582-7.].
). From a subset of amino acids within 5 Å of the bound ligand, Lys175, Cys176, and Tyr273 appeared to have the most contact with the ligand. It was reasonable to assume that decreasing the size of the side chain could open up the tunnel and facilitate more rapid ligand entry and binding. Additional evidence for the importance of positions 176 and 273 came from a previous report on the engineering of a DhaA variant containing C176Y and Y273F. This double mutant could bind more efficiently to 1,2,3-trichloropropane, suggesting a role for these residues in positioning incoming haloalkane substrates for efficient nucleophilic attack [35 Bosma T, Damborsky J, Stucki G, Janssen DB. Biodegradation of
1,2,3-trichloropropane through directed evolution and heterologous
expression of a haloalkane dehalogenase gene Appl Environ Microbiol
2002; 68(7
): 3582-7.].

We carried out saturation mutagenesis individually at codons 175, 176 and 273 and also at both position 175 and 176 in a combined mutagenesis reaction. Each library (fusions to GST) was screened in bacterial lysates for variants with improved binding rates. The most beneficial substitutions (K175M/C176G and Y273L) were combined to create the variant, HT2 (K175M/C176G/Y273L). Kinetic analysis indicated the mutations were additive in nature (Fig. 5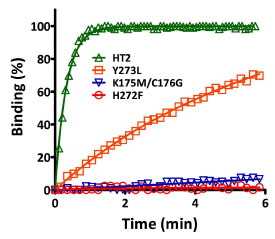 ), and HT2 displayed binding kinetics that were more comparable to the interaction between streptavidin and biotin [21 Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein
labeling technology for cell imaging and protein analysis ACS
Chem Biol 2008; 3(6
): 373-82., 34 Qureshi MH, Yeung JC, Wu S-C, Wong S-L. Development and
characterization of a series of soluble tetrameric and monomeric
streptavidin muteins with differential biotin binding affinities J
Biol Chem 2001; 276(49
): 46422-8.]. Apparent second-order rate constants for the reactions between HT2 and the FAM- and TMR-ligands were 1.6 x 104 and 3.0 x 106 M-1sec-1, respectively. These values indicated binding kinetic improvements of ~10,000-fold for the FAM-ligand and ~40,000-fold for the TMR-ligand.
), and HT2 displayed binding kinetics that were more comparable to the interaction between streptavidin and biotin [21 Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein
labeling technology for cell imaging and protein analysis ACS
Chem Biol 2008; 3(6
): 373-82., 34 Qureshi MH, Yeung JC, Wu S-C, Wong S-L. Development and
characterization of a series of soluble tetrameric and monomeric
streptavidin muteins with differential biotin binding affinities J
Biol Chem 2001; 276(49
): 46422-8.]. Apparent second-order rate constants for the reactions between HT2 and the FAM- and TMR-ligands were 1.6 x 104 and 3.0 x 106 M-1sec-1, respectively. These values indicated binding kinetic improvements of ~10,000-fold for the FAM-ligand and ~40,000-fold for the TMR-ligand.

Note that because of the hydrophobic nature of TMR we were concerned that non-specific interactions may have been causing artifactual FP signals. The amount of CHAPS in these reactions should prevent such interactions, but as a precaution we repeated the kinetic analysis using SDS-PAGE and quantitative fluorescence scanning. Any products of non-specific binding between protein and ligand should have been eliminated by the denaturing conditions of this assay format, yet we calculated similar kinetic parameters for both ligands using this alternative method. This suggested that the FP assay was not susceptible to this type of artifact and was therefore a reliable approach for accessing the binding rate of HT2 and subsequently derived variants.
To understand the role of the amino acid substitutions in the improved binding kinetics, we created a 3-dimensional structure model of HT2 in the absence of bound ligand and compared it to a similar model created for H272F (Fig. 6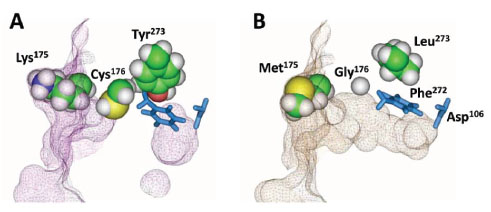 ). H272F (panel A) showed a distinct tunnel entrance at the protein surface near Lys175 and a large cavity near the catalytic triad, separated by a significant constriction in the tunnel near Cys176 and extending to Tyr273. In contrast, the tunnel for HT2 (panel B) displayed a continuously open structure. The model indicates the K175M substitution did not play a significant role in the opening of the tunnel, suggesting its role involved more subtle steric effects or perhaps the removal of charge. The model also suggests that substitutions at positions 176 and 273 allowed repositioning of adjacent side chains. In the H272F model the Phe272 side chain protruded into the tunnel in the absence of ligand, requiring a ~45o rotation to enable ligand entry. In HT2, the Phe272 side chain appears to already be in a position optimal for ligand entry. Furthermore, a slightly different view of the model (not shown) indicated that the proposed structurally important Glu130 (Fig. 1) was pushed away from the tunnel by Phe272 in H272F, while this residue was unaffected in HT2.
). H272F (panel A) showed a distinct tunnel entrance at the protein surface near Lys175 and a large cavity near the catalytic triad, separated by a significant constriction in the tunnel near Cys176 and extending to Tyr273. In contrast, the tunnel for HT2 (panel B) displayed a continuously open structure. The model indicates the K175M substitution did not play a significant role in the opening of the tunnel, suggesting its role involved more subtle steric effects or perhaps the removal of charge. The model also suggests that substitutions at positions 176 and 273 allowed repositioning of adjacent side chains. In the H272F model the Phe272 side chain protruded into the tunnel in the absence of ligand, requiring a ~45o rotation to enable ligand entry. In HT2, the Phe272 side chain appears to already be in a position optimal for ligand entry. Furthermore, a slightly different view of the model (not shown) indicated that the proposed structurally important Glu130 (Fig. 1) was pushed away from the tunnel by Phe272 in H272F, while this residue was unaffected in HT2.

We examined HT2 as a tool for both cellular imaging and protein immobilization, and found that it enabled both applications. CHO-K1 cells expressing HT2 that were treated with the TMR-ligand were significantly brighter than those expressing the parental variant, H272F (Fig. S2A-C). Furthermore, less ligand and shorter incubation times were required to efficiently label cells, thereby eliminating the need for stringent washing to remove unbound ligand [33 Los GV, Wood K. The HaloTag: a novel technology for cell imaging and protein analysis Methods Mol Biol 2007; 356: 195-208.]. We also found that HT2 could be immobilized to a chloroalkane surface (i.e. streptavidin microtiter plate coated with a biotinylated chloroalkane ligand; PEG Biotin-ligand) (Fig. S2D), suggesting its potential utility for isolating proteins from complex samples.
Engineering HT2 for Structural Compatibility with Fusion Partners
It is essential that a protein tag be structurally compatible with fused target proteins. To examine HT2 for this feature we fused it to the N- and C-termini of a variety of proteins and measured the production of protein. The fusions generally expressed poorly in both cell-free systems and E. coli. This was exemplified by fusions to humanized Renilla luciferase (Rluc) (Fig. 7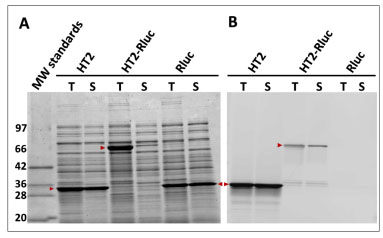 ). This fusion (HT2-Rluc) and both of the individual proteins were overexpressed in E. coli and both crude and soluble fractions of the lysates were labeled to completion (20 µM TMR-ligand, 1 h, 25 oC). Samples were analyzed by SDS-PAGE and the resulting gel scanned and quantitated for fluorescence. This allowed us to assess the amount of total (T) and soluble (S) protein using the Simply-Blue-stained gel image (panel A) and the amount of functional protein in both fractions using the fluorescence image (panel B). Both HT2 and Rluc were produced efficiently as soluble proteins. HT2-Rluc also expressed well but was largely insoluble. Fluorescence labeling of HT2-Rluc was relatively low, indicating a majority of the fusion was non-functional. Loss in functionality for the fusion was further confirmed by the significant decrease (~30-fold) in Rluc luminescence observed for HT2-Rluc compared to Rluc alone (data not shown). The general trend represented by this result indicated a limitation for HT2 as a fusion tag.
). This fusion (HT2-Rluc) and both of the individual proteins were overexpressed in E. coli and both crude and soluble fractions of the lysates were labeled to completion (20 µM TMR-ligand, 1 h, 25 oC). Samples were analyzed by SDS-PAGE and the resulting gel scanned and quantitated for fluorescence. This allowed us to assess the amount of total (T) and soluble (S) protein using the Simply-Blue-stained gel image (panel A) and the amount of functional protein in both fractions using the fluorescence image (panel B). Both HT2 and Rluc were produced efficiently as soluble proteins. HT2-Rluc also expressed well but was largely insoluble. Fluorescence labeling of HT2-Rluc was relatively low, indicating a majority of the fusion was non-functional. Loss in functionality for the fusion was further confirmed by the significant decrease (~30-fold) in Rluc luminescence observed for HT2-Rluc compared to Rluc alone (data not shown). The general trend represented by this result indicated a limitation for HT2 as a fusion tag.

We hypothesized that the underlying cause of poor expression was inefficient folding caused by a non-compatible HT2 structure, and that this could be resolved by engineering greater structural stability into the tag. We first considered that perhaps Phe272 was a liability to HT2, because unlike the His in the native enzyme Phe cannot form a stabilizing hydrogen bond with Glu130 [14 Newman J, Peat TS, Richard R, et al. Haloalkane dehalogenases:
structure of a Rhodococcus enzyme Biochemistry 1999; 38(49
): 16105-4.] (Fig. 1). We predicted that replacement of the Phe with a residue that could form a bond to Glu130 would stabilize HT2. Because the adjacent residue at position 273 was critical to binding kinetics, we tried to identify the optimal pair of amino acid substitutions for these two positions. A library of all amino acid combinations for these two sites was constructed and screened in the context of a C-terminal fusion partner (chloramphenicol acetyltransferase). We used an FP-based assay (FAM-ligand) for improved expression to screen the library in bacterial lysates. Each improved variant contained Asn272, a residue that theoretically should be able to hydrogen bond to Glu130 (A computational structure analysis of Asn272 can be found in the
Supplementary Material; Fig.
S3). The improved variants also contained either Leu (NL) or Phe (NF) at position 273.
Further characterization of NL and NF (using elevated temperature inductions at 30 oC for increased stringency) revealed that both produced more soluble and functional protein (NL was ~10-fold improved; NF was ~5-fold improved). NL displayed ~4-fold slower binding kinetics than HT2 (FAM-ligand), while NF offered subtle but further improved kinetics over HT2. Because NF provided improved expression without sacrificing binding kinetics, it was considered to be the superior variant and more appropriate template for further optimization. A second approach to stabilizing HT2 involved testing mutations that were previously shown to improve the thermal stability of DhaA [36 Gray KA, Richardson TH, Kretz K, et al. Rapid evolution of reversible denaturation and elevated melting temperature in a microbial haloalkane dehalogenase Adv Synth Catal 2001; 343(6-7 ): 607-17.]. We found that D78G provided a modest enhancement to expression when combined with NF, and the resulting variant (GNF) was used as the template for subsequent molecular optimization.
To further improve the stability and expression of GNF we created a random library of mutations across the entire coding sequence using error-prone PCR (Note prior to making this library the nucleotide sequence of GNF was optimized by the removal of rarely used codons in both E. coli and human genes). We screened ~26,000 variants as N-terminal fusions to Rluc in bacterial lysates and identified six variants of interest, each containing a single amino acid substitution. Five of the substitutions (S58T, A155T, A224E, P291S, and A292T) provided enhanced expression (1.2–1.5-fold), while a sixth (A172T) was neutral for expression but provided faster FAM-ligand binding kinetics. The six substitutions were combined and the resulting variant (HT2.1) produced 2.5-fold more functional fusion protein than GNF (Fig. 8 ). Because Leu273 was previously shown to improve expression in the context of Asn272, we introduced it to HT2.1 to give the variant, HT3. HT3 produced 6-fold more soluble and functional protein than GNF and displayed ligand binding kinetics comparable to HT2. Similar magnitudes of improvement were observed when HT3 was fused to the N-termini of firefly luciferase (Fluc) and Id. The presence of a ~34 kDa protein in the fluorescence scans may be due to proteolytic cleavage of the linker sequence between the fused proteins to produce free HT3. This product is not apparent in the SimplyBlue-stained gels, as it is below the limit of detection.
). Because Leu273 was previously shown to improve expression in the context of Asn272, we introduced it to HT2.1 to give the variant, HT3. HT3 produced 6-fold more soluble and functional protein than GNF and displayed ligand binding kinetics comparable to HT2. Similar magnitudes of improvement were observed when HT3 was fused to the N-termini of firefly luciferase (Fluc) and Id. The presence of a ~34 kDa protein in the fluorescence scans may be due to proteolytic cleavage of the linker sequence between the fused proteins to produce free HT3. This product is not apparent in the SimplyBlue-stained gels, as it is below the limit of detection.

HT3 was further examined by fusing it to the C-termini of Rluc, Fluc, and Id. Although HT3 was beneficial to expression in this context, the magnitude of the improvements was not as significant as with N-terminal fusions. This was somewhat expected because the tag should have reduced ability to effect folding when it is synthesized subsequent to the target protein. Although expression tags for E. coli are generally placed on the N-termini of fusion partners, we wanted to optimize HT3 as a general tag, including its use as the C-terminal partner in a fusion protein. We therefore carried out additional optimization of the tag in the context of C-terminal fusions to Rluc, Fluc, and Id. The purpose of screening the library in the context of multiple partners was to guide our selection of beneficial mutations towards those providing general expression or stability improvements to HT3 rather than mutations that were specific to a particular fusion partner. Id and Fluc were chosen for this purpose to increase the stringency of the screen, as they were both poorly-expressed in E. coli even in the absence of a tag (data not shown).
We screened 48,000 variants from the three libraries using the FAM-ligand FP assay and validated the most improved variants as before. Many of the best mutations were common to all three libraries, suggesting their impact was general in nature. The beneficial mutations were also examined as N-terminal fusions to Id, Fluc, and Rluc, and any that were detrimental to expression or binding kinetics in this context were eliminated from further consideration. Ultimately, nine substitutions (L47V, Y87F, L88M, C128F, E160K, K195N, N227D, E257K, and T264A) were identified as providing the most improved expression of soluble and functional protein (with no impact on binding kinetics) and one substitution (A167V) that provided further enhanced ligand binding kinetics. A composite of all ten mutations (HT6) was examined for expression in E. coli as both N- and C-terminal fusions to Id, Fluc, and Rluc and found to produce higher levels of soluble and functional fusion protein in both orientations with all three partners (Fig 9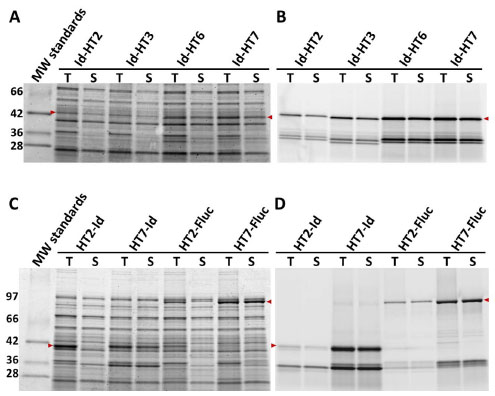 ). This variant was ultimately also shown to display significantly faster ligand binding kinetics than HT3 in the absence of a fusion partner (Table 1).
). This variant was ultimately also shown to display significantly faster ligand binding kinetics than HT3 in the absence of a fusion partner (Table 1).


Apparent Rate Constants (k)a for Binding Reactions between H272F-based Variants and Haloalkane Ligands as Determined by FP
Throughout the optimization process, beneficial substitutions were frequently found near the C-terminus of the tag (e.g. positions 291 and 292). Both the crystal structure of DhaA [14 Newman J, Peat TS, Richard R, et al. Haloalkane dehalogenases:
structure of a Rhodococcus enzyme Biochemistry 1999; 38(49
): 16105-4.] and our own models of different variants indicated an α -helix at the C-terminus originating in close proximity to the base of the ligand binding tunnel. This suggested that any structural perturbation of the helix imposed by a fusion partner could be transmitted to this critical region of the tag known to play an important role in both stability and ligand binding kinetics. We therefore attempted to optimize the helix by random amino acid substitution at positions 291–293 and by introducing random two-residue extensions (i.e. positions 294–295). The library was fused to the C-terminus of Id and screened as E. coli lysates for improved expression of soluble functional protein by labeling to completion with the TMR-ligand and analyzing by SDS-PAGE and fluorescence scanning. We identified an improved variant (HT7) with a C-terminal (positions 291–297) sequence of Ser-Thr-Leu-Glu-Ile-Ser-Gly (Note the terminal Ser-Gly were present as part of an AccIII restriction site used for cloning.). HT7 was verified to provide improved or neutral expression in multiple N- and C-terminal fusion contexts. As a C-terminal tag to Id it provided 7-fold more functional full length fusion protein compared to the original HT2 (Fig. 9A,B). As an N-terminal tag to Id and Fluc it was improved by ~80- and 10-fold, respectively (Fig. 9C,D).
The HT7 variant represents the final evolved version of the tag and is referred to generally as HaloTag. Additional information, including a summary of the mutations and a structure model highlighting the location of the amino substitutions, can be found in the Supplementary Material (Table S1, Fig. S4).
Binding Kinetics–HT2, HT3, HT6 and HT7
To characterize the ligand binding kinetics for HT7, it was purified as a GST fusion and then the GST tag was removed by proteolytic cleavage (TEV). HT2, HT3, and HT6 were purified in the same manner so that the four variants could be directly compared. We measured the kinetics of labeling (FAM- and TMR-ligands) and calculated apparent rate constants as before (Table 1). The binding kinetics for both ligands was further improved going from HT3 to HT7. Note that the apparent second order rate constant for binding of the TMR-ligand to HT7, 1.9 x 107 M-1sec-1, was over 2-fold higher than the value previously calculated for the reaction between biotin (TMR-biotin) and streptavidin [21 Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis ACS Chem Biol 2008; 3(6 ): 373-82.].
Linker Optimization
In addition to optimizing the tag we engineered peptide linkers to connect HT7 to either the N- or C-terminus of target proteins. The linkers were optimized for fusion stability and efficient proteolytic (TEV protease-mediated) release of target protein. Additional details on the linkers can be found in the Supplementary Material (Fig. S5, Table S2)
HT7 Improves the Expression of Rluc
To further understand the magnitude of the overall benefit provided by HT7 as an N-terminal tag in combination with the optimized linker, we revisited the experiment summarized in Fig. (7). When overexpressed in E. coli KRX, HT7-Rluc provided an equivalent amount of soluble total protein compared to Rluc alone (Fig. 10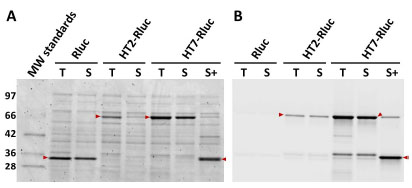 ), and ~25-fold more soluble and functional fusion protein compared to HT2-Rluc (linker N-3; see the
Supplementary Material). We measured Rluc activity for these lysates and the luminescence for HT7-Rluc was also improved ~25-fold, to the extent that it was now ~50% as bright as non-tagged Rluc (compared to only ~3% for HT2-Rluc). In addition, we incubated the HT7-Rluc lysates with TEV protease and observed efficient cleavage (~90%) of the fusion. The Rluc activity was also measured for this sample, and found to be unchanged compared to the non-cleaved sample. This indicated removal of HT7 from Rluc did not impact the functionality of the luciferase.
), and ~25-fold more soluble and functional fusion protein compared to HT2-Rluc (linker N-3; see the
Supplementary Material). We measured Rluc activity for these lysates and the luminescence for HT7-Rluc was also improved ~25-fold, to the extent that it was now ~50% as bright as non-tagged Rluc (compared to only ~3% for HT2-Rluc). In addition, we incubated the HT7-Rluc lysates with TEV protease and observed efficient cleavage (~90%) of the fusion. The Rluc activity was also measured for this sample, and found to be unchanged compared to the non-cleaved sample. This indicated removal of HT7 from Rluc did not impact the functionality of the luciferase.
 |
Fig. (10) Expression of HT7-Rluc with optimized linker. HT7-Rluc (linker N-HT7; see the Supplementary Material), HT2-Rluc (linker N-3; see Supplementary Material), and Rluc were overexpressed in E. coli KRX at 25 °C and then lysate fractions containing total (T) or soluble (S) protein were labeled to completion with the TMR-ligand and resolved by SDS-PAGE. TMR-labeled HT7-Rluc was also incubated with TEV protease (S+) for 30 min at 30 °C. Gels were imaged for both total protein (SimplyBlue, panel A) and the amount of functional fusion (TMR fluorescence, panel B). Relative amounts of soluble and functional (TMR-labeled) protein (full length and proteolytically cleaved) could be quantitated from the fluorescence scan (Eex/Eem=532/580 nm). Overlaid arrows indicate bands of interest. Note the ~34 kDa band in panel B which presumably represents truncation of the fusion. |
To investigate whether the benefits provided by HT7 could be realized in other expression systems (e.g. cell-free systems, mammalian cells) we fused it to a variety of different partners, as both N- and C-terminal tags in vectors appropriate for each expression system. In general we observed the same pattern of expression improvements found in E. coli. Please see the Supplementary Material for specific examples as well as a summary of improved levels of protein production that have been observed using alternative expression systems (Fig. S6, S7; Tables S3, S4).
Further Characterization of HT7
To investigate the stoichiometry of the reaction between HT7 and ligand we used the mass spectrometry-based approach already described for H272F. As was the case for H272F, the product of the binding reaction had a molecular mass consistent with a single binding event. In addition, trypsin digestion of this same product and mass analysis of the resulting peptides indicated that the mass gain for the labeled protein was localized to the appropriate 31-amino acid fragment containing the reactive nucleophile (Asp106). We further examined the stability of the ester bond-based attachment by exposing TMR-labeled HT7 for 30 min to a wide range of temperature and pH conditions, and then analyzed the protein by SDS-PAGE and fluorescence scanning. The stability of the TMR attachment was unaffected by elevated temperature in the presence of SDS, and the bond was resistant to hydrolysis at either alkaline or acidic pH (Fig. S8). HT7 was further analyzed using gel permeation chromatography, and like HT2 it was shown to be monomeric (data not shown).
HT7 structural stability was further examined by exposing purified protein to elevated temperature and chemical denaturants. We used circular dichroism analysis to ascertain changes in secondary structure as a function of temperature by monitoring changes in ellipticity at 224 nm (Fig. 11A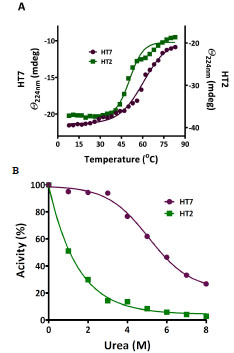 ). The results indicated denaturation temperatures (Tm) of 61 oC for HT7 and 51 oC for HT2. In addition to the melting analysis, a FAM-ligand binding assay was carried out on HT7 and HT2 following exposure to elevated temperature, and similar Tm values were observed (Fig.
S9). We also examined the effect of urea on the stability of HT2 and HT7 using pulse proteolysis [26 Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative
determination of protein stability and ligand binding Nat
Methods 2005; 2(3
): 207-12.]. TMR-labeled protein was exposed overnight at 22 oC to the denaturants and analyzed for proper folding based on sensitivity to proteolytic cleavage by thermolysin. We defined 100% properly folded protein as the degree of cleavage (as determined by SDS-PAGE and fluorescence scanning) observed for each protein in the absence of urea, and calculated the amount of properly folded protein following exposure to the denaturant (Fig. 11B). HT2 was sensitive to all concentrations of urea tested, while HT7 maintained significant activity even after exposure to urea concentrations as high as 6 M. Guanidinium was also tested as a denaturant, and similar results were obtained (data not shown). We also examined the impact of a pH, NaCl, and variety of common detergents on HT7 ligand binding. A summary of these experiments can be found in the
Supplementary Material (Fig.
S10, Tables
S5,
S6).
). The results indicated denaturation temperatures (Tm) of 61 oC for HT7 and 51 oC for HT2. In addition to the melting analysis, a FAM-ligand binding assay was carried out on HT7 and HT2 following exposure to elevated temperature, and similar Tm values were observed (Fig.
S9). We also examined the effect of urea on the stability of HT2 and HT7 using pulse proteolysis [26 Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative
determination of protein stability and ligand binding Nat
Methods 2005; 2(3
): 207-12.]. TMR-labeled protein was exposed overnight at 22 oC to the denaturants and analyzed for proper folding based on sensitivity to proteolytic cleavage by thermolysin. We defined 100% properly folded protein as the degree of cleavage (as determined by SDS-PAGE and fluorescence scanning) observed for each protein in the absence of urea, and calculated the amount of properly folded protein following exposure to the denaturant (Fig. 11B). HT2 was sensitive to all concentrations of urea tested, while HT7 maintained significant activity even after exposure to urea concentrations as high as 6 M. Guanidinium was also tested as a denaturant, and similar results were obtained (data not shown). We also examined the impact of a pH, NaCl, and variety of common detergents on HT7 ligand binding. A summary of these experiments can be found in the
Supplementary Material (Fig.
S10, Tables
S5,
S6).
 |
Fig. (11) Structural stability of HT7 and HT2. A. Temperature dependence of CD signal at 224 nm. Denaturation temperatures (Tm) were determined by fitting the data to a simple two-state transition model. B. Effect of urea on ligand binding activity. Proteins were exposed to urea for 16 h at 25 °C and the amount of properly folded protein remaining (relative to no treatment) was calculated based on sensitivity to thermolysin-induced proteolysis [26 Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding Nat Methods 2005; 2(3 ): 207-12.]. |
Applications for HT7 (Isolation of Functional Ribosomes)
HT7 has been used successfully in a variety of applications including cellular imaging [37 Kosaka N, Ogawa M, Choyke PL, et al. In vivo stable tumor-specific
painting in various colors using dehalogenase-based protein-
tag fluorescent ligands Bioconjug Chem 2009; 20(7
): 1367-74., 38 Lee H-lD, Lord SJ, Iwanaga S, Zhan K, Xie H, Williams JC, et al.
Superresolution imaging of targeted proteins in fixed and living
cells using photoactivatable organic fluorophores J Am Chem Soc
2010; 132(43
): 15099-01.], expression and purification of difficult proteins [39 Chumanov RS, Kuhn PA, Xu W, Burgess RR. Expression and
purification of full-length mouse CARM1 from transiently transfected
HEK293T cells using HaloTag technology Protein Expression
Purif 2010; 76(2
): 145-53.-43 Ohana RF, Hurst R, Vidugiriene J, Slater MR, Wood KV, Urh M.
HaloTag-based purification of functional human kinases from
mammalian cells Protein Expression Purif 2011; 76(2
): 154-64.], and the interrogation of protein:DNA and protein:protein interactions [44 Daniels DL, Mendez J, Mosley AL, et al. Examining the complexity
of human RNA polymerase complexes using HaloTag technology
coupled to label free quantitative proteomics J Proteome Res
2012; 11: 564-75.-48 Gallo S, Beugnet A, Biffo S. Tagging of functional ribosomes in
living cells by HaloTag® technology In Vitro Cellular & Developmental
Biology - Animal 2011; 47(2
): 132-8.]. To determine if we could efficiently isolate, measure activity, and monitor in vivo localization of a macromolecular machine complex, we appended HT7 to the C-terminus of RPS9, a component of the small 40S ribosomal subunit. RPS9-HT7 was transiently transfected and expressed in HEK-293T cells, and after lysis, captured along with interacting protein partners using sepharose beads coated with HT7 ligands (HaloLink™ Resin). Isolated RPS9 complexes were released from the resin (by TEV protease cleavage) and upon analysis by SDS-PAGE and silver staining shown to contain a significant number of distinct bands (Fig. 12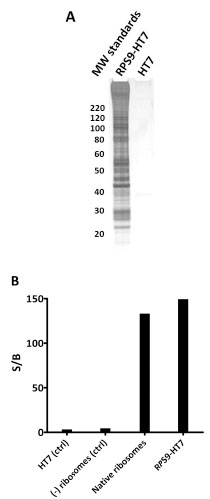 ). Mass spectrometry analysis revealed nearly complete capture of the 40S and 60S subunits, indicating efficient isolation of intact 80S ribosomes (Fig. 12A and Table
S7). The detection of additional initiation, translation, and polyA-associated proteins suggested capture of actively translating polysomes. To determine whether ribosomes isolated using RPS9-HT7 were functional for in vitro translation we isolated them from HEK-293T cells stably expressing Fluc mRNA and measured their ability to translate ribosome-bound luciferase mRNA. Luciferase activity was detected, and at a level comparable to that from a reaction between commercially available ribosomes and Fluc mRNA (Fig. 12B). These data combined with the mass data indicate the ribosomes captured using HT7 were fully formed 80S particles and functional for in vitro translation.
). Mass spectrometry analysis revealed nearly complete capture of the 40S and 60S subunits, indicating efficient isolation of intact 80S ribosomes (Fig. 12A and Table
S7). The detection of additional initiation, translation, and polyA-associated proteins suggested capture of actively translating polysomes. To determine whether ribosomes isolated using RPS9-HT7 were functional for in vitro translation we isolated them from HEK-293T cells stably expressing Fluc mRNA and measured their ability to translate ribosome-bound luciferase mRNA. Luciferase activity was detected, and at a level comparable to that from a reaction between commercially available ribosomes and Fluc mRNA (Fig. 12B). These data combined with the mass data indicate the ribosomes captured using HT7 were fully formed 80S particles and functional for in vitro translation.
 |
Fig. (12) Capture of intact 80S ribosome from HEK-293T cells using RPS9-HT7. A. Overexpressed RPS9-HT7 (or HT7 alone, control) was captured to HaloLink resin and treated with TEV protease to release RPS9 and its interacting partners. The eluted samples were analyzed by SDS-PAGE and silver staining. Mass analysis of the same samples verified the following was present: 31 of 33 40S proteins, 42 of 50 60S proteins, 2 poly-A binding proteins, 1 GNF exchange protein, 9 nuclear ribonucleoproteins, 2 initiation factors, 2 elongation factors, and 2 splicing factors. For a complete list see Table S7. BIn vitro luciferase translation assay showing activity of ribosomes isolated via RPS9-HT7. RPS9-HT7 was transiently expressed in HEK-293T cells stably expressing Fluc mRNA. Ribosomes were isolated via RPS9-HT7 and released using TEV protease. HT7 alone and untransfected cells were processed in the same manner as negative controls. Signal to background calculations indicated the generation of active luciferase from the RPS9- HT7 complex isolation but not from the negative controls. Commercially available native ribosomes, included as a positive control, were also able to generate active luciferase in vitro. |
To monitor protein localization and cellular trafficking of ribosomes, a stable U2OS cell line expressing RPS9-HT7 was analyzed using two fluorescent HT7 ligands in pulse labeling experiments (Fig. 13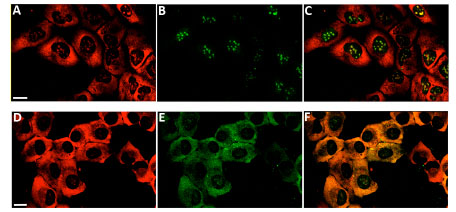 ). Initial labeling of RPS9-HT7 with the TMR-ligand (panels A, D) showed the majority of localization was to the cytoplasm with some signal in the nucleoli where ribosome assembly occurs. Pulse labeling of new populations of RPS9-HT7 with the Oregon Green-ligand showed strict nucleolar localization at 3 h (panel B), yet by 24 h (panel E) RPS9-HT7 was found in both the cytoplasm and nucleoli. Panels C and F are overlays of panels A,B and D,E, respectively. These results demonstrate the cellular pathway of RPS9-HT7 followed that of expected ribosome subunits, i.e. assembly in the nucleoli and then translocation to the cytoplasm.
). Initial labeling of RPS9-HT7 with the TMR-ligand (panels A, D) showed the majority of localization was to the cytoplasm with some signal in the nucleoli where ribosome assembly occurs. Pulse labeling of new populations of RPS9-HT7 with the Oregon Green-ligand showed strict nucleolar localization at 3 h (panel B), yet by 24 h (panel E) RPS9-HT7 was found in both the cytoplasm and nucleoli. Panels C and F are overlays of panels A,B and D,E, respectively. These results demonstrate the cellular pathway of RPS9-HT7 followed that of expected ribosome subunits, i.e. assembly in the nucleoli and then translocation to the cytoplasm.

DISCUSSION
Here we describe the development of HT7, a genetic fusion tag that can be used to efficiently label and capture proteins of interest for a variety of applications. HT7 was engineered to possess a combination of desirable properties not found for many commonly used affinity/epitope tags. It binds with high specificity to a synthetic ligand and forms a covalent attachment that is stable enough to withstand rigorous washing. Binding is highly efficient because the interaction is rapid and essentially irreversible. In contrast, common affinity tags are equilibrium-based, and as a result are susceptible to inefficient binding when present at low concentrations. The binding efficiency of affinity tags can also be compromised by washing, as the removal of unbound tag from a sample causes bound tag to dissociate upon re-equilibration. Although epitope tags bind to antibodies with high affinity and specificity, binding capacity can be limited by steric effects [49 Einhauer A, Jungbauer A. The FLAG peptide, a versatile fusion tag for the purification of recombinant proteins J Biochem Biophys Methods 2001; 49(1-3 ): 455-65.] or surface decay [50 Joss L, Morton TA, Doyle ML, Myszka DG. Interpreting kinetic rate constants from optical biosensor data recorded on a decaying surface Anal Biochem 1998; 261(2 ): 203-10.]. The binding ligand for HT7 was designed to carry different functionalities (e.g. fluorophores, attachments to solid supports), allowing the same genetic construct to be used for multiple applications. Moreover, HT7 has been structurally optimized through molecular evolution to provide efficient production of functionally competent fusion proteins from a variety of expression hosts.
DhaA was an appropriate starting point for the molecular evolution of a fusion tag because it forms a transient covalent attachment to its native substrates, which can be trapped by introducing a single amino acid substitution to its catalytic pocket [14 Newman J, Peat TS, Richard R, et al. Haloalkane dehalogenases: structure of a Rhodococcus enzyme Biochemistry 1999; 38(49 ): 16105-4., 16 Janssen DB. Evolving haloalkane dehalogenases Curr Opin Chem Biol 2004; 8(2 ): 150-9., 19 Pieters RJ, Fennema M, Kellogg RM, Janssen DB. Design and synthesis of reagents for phage display screening of dehalogenases Bioorg Med Chem Lett 1999; 9(2 ): 161-6.]. This specialized hydrolase was also attractive as a potential tag because it is small, monomeric, does not require co-factors, metal ions, or post-translational modifications, and is not subject to product inhibition [9 Kulakova AN, Larkin MJ, Kulakov LA. The plasmid-located haloalkane dehalogenase gene from Rhodococcus rhodochrous NCIMB 13064 Microbiology 1997; 143(Pt 1): 109-5., 14 Newman J, Peat TS, Richard R, et al. Haloalkane dehalogenases: structure of a Rhodococcus enzyme Biochemistry 1999; 38(49 ): 16105-4., 51 Stafford TM. Haloalkane degradation by Rhodococcus rhodochrous NCZMB 13064 [PhD Dissertation]. : The Queen's University of Belfast1993.]. Furthermore it is absent from eukaryotes and many prokaryotes (including E. coli), thereby minimizing the risk of background interactions between haloalkane-based ligands and common experimental hosts. It efficiently processes primary chloroalkanes, which are chemically simple and easily customizable by straightforward synthetic chemistries. Finally, DhaA has broad substrate specificity compared to other dehalogenases [7 Schindler JF, Naranjo PA, Honaberger DA, et al. Haloalkane dehalogenases: steady-state kinetics and halide inhibition Biochemistry 1999; 38(18 ): 5772-8., 12 Yokota T, Omori T, Kodama T. Purification and properties of haloalkane dehalogenase from Corynebacterium sp. strain m15-3 J Bacteriol 1987; 169(9 ): 4049-54., 13 Bosma T, Kruizinga E, de Bruin EJ, Poelarends GJ, Janssen DB. Utilization of trihalogenated propanes by Agrobacterium radiobacter AD1 through heterologous expression of the haloalkane dehalogenase from Rhodococcus sp. strain M15-3 Appl Environ Microbiol 1999; 65(10 ): 4575-81.], presumably because of a wider and deeper active site cavity [14 Newman J, Peat TS, Richard R, et al. Haloalkane dehalogenases: structure of a Rhodococcus enzyme Biochemistry 1999; 38(49 ): 16105-4.]. This suggested a greater likelihood that it could accept modified haloalkanes containing spacer segments and functional moieties (e.g. fluorophores, biotin, or capture surfaces) as substrates or eventual binding ligands.
The optimal structure for the chloroalkane binding ligand was empirically determined by testing different spacers between the chlorine and the functionality (FAM, TMR). The optimal spacer was 17 Å, consistent with our structure model-based prediction that the depth of the binding tunnel was 15 Å. Providing length was not the only role of the linker. It is possible that the polyethylene glycol units provided solubility and as well as a more rigid molecular structure that facilitated entrance to the binding tunnel. The glycol oxygens may also facilitate ligand penetration of the partially hydrophilic binding tunnel. The tolerance of the polyethylene glycol units by DhaA was also attractive from an application standpoint, as the glycol oxygens may improve cell permeability. Furthermore, the presence of related glycol moieties in solid surface matrices is known to reduce non-specific protein interactions [52 Pale-Grosdemange C, Simon ES, Prime KL, Whitesides GM. Formation of self-assembled monolayers by chemisorption of derivatives of oligo(ethylene glycol) of structure HS(CH2)11(OCH2CH2) mOH on gold J Am Chem Soc 1991; 113(1 ): 12-20.]. Finally, these ligands showed neither cytotoxicity nor any impact on cell morphology when applied to cells at relevant concentrations [21 Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis ACS Chem Biol 2008; 3(6 ): 373-82.].
The covalent intermediate formed between DhaA and substrate was originally trapped by replacing the catalytic base (His272) with Phe. Through random mutagenesis we found that Asn was the preferred residue at this position for structural stability, presumably because of improved space filling and the ability to form a stabilizing hydrogen bond with Glu130. The trapped ester bond between HT7 and ligand was resistant to hydrolysis at elevated temperature, in the presence of SDS, and across a broad pH range. The stability of the attachment was presumably due to its location in a microenvironment deep within the ligand access tunnel where it is difficult for hydrolysis to occur. The protected location of the bond, combined with the surrounding hydrophobicity of the protein and bound ligand, may serve to exclude water from the immediate vicinity of the bond [16 Janssen DB. Evolving haloalkane dehalogenases Curr Opin Chem Biol 2004; 8(2 ): 150-9.].
Although H272F formed a stable attachment to ligands, its utility as a labeling or capture tool was limited by slow binding kinetics. DhaA naturally evolved to recognize substrates of smaller size and lower complexity than our ligands [53 Janssen DB, Dinkla IJT, Poelarends GJ, Terpstra P. Bacterial degradation of xenobiotic compounds: evolution and distribution of novel enzyme activities Environ Microbiol 2005; 7(12 ): 1868-82.], and the rate limiting step in catalysis is product release [17 Bosma T, Pikkemaat MG, Kingma J, Dijk J, Janssen DB. Steadystate and pre-steady-state kinetic analysis of halopropane conversion by a rhodococcus haloalkane dehalogenase Biochemistry 2003; 42(26 ): 8047-53.]. The absence of any natural selective pressure on DhaA to improve its initial binding rate added to its appeal as a target for laboratory molecular evolution. The binding kinetics for H272F were improved dramatically (10,000–40,000-fold depending on the ligand) by randomly mutating critical sites in the binding tunnel and then combining beneficial substitutions. A structure model of the resulting variant, HT2, indicated a wider and more continuous binding pocket compared to H272F. When benchmarked against streptavidin and biotin, one of the fastest known biomolecular interactions [34 Qureshi MH, Yeung JC, Wu S-C, Wong S-L. Development and characterization of a series of soluble tetrameric and monomeric streptavidin muteins with differential biotin binding affinities J Biol Chem 2001; 276(49 ): 46422-8.], the reaction between HT2 and the TMR-ligand proceeded with similar kinetics [21 Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis ACS Chem Biol 2008; 3(6 ): 373-82.]. Faster binding by the TMR-ligand compared to the FAM-ligand indicates that although the fluorophore was distant from the chlorine it still played a role in binding kinetics. The difference between ligands may have been due to electrostatics. The entrance to the binding tunnel is located in a patch of negative charge, which could perturb interactions with the negatively charged FAM-ligand. In contrast, the more hydrophobic nature of the TMR-ligand may contribute to faster binding via interactions with non-polar amino acid side chains at the tunnel entrance. Attempts to validate these predictions using our structure model led to inconclusive evidence that residues near the tunnel entrance played a role in the kinetic differences between the two ligands.
We demonstrated here that HT2 was a capable tag for cellular imaging and protein immobilization. Additional examples of the tag’s utility include the imaging and characterization of p65 nuclear translocation [21 Los GV, Encell LP, McDougall MG, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis ACS Chem Biol 2008; 3(6 ): 373-82.], hydrophobic tagging for the study of protein degradation [54 Neklesa TK, Tae HS, Schneekloth AR, et al. Small-molecule hydrophobic tagging-induced degradation of HaloTag fusion proteins Nat Chem Biol 2011; 7(8 ): 538-43.], and the conjugation of bioluminescent enzymes to quantum dots or the labeling of cells with quantum dots for optical imaging [55 Zhang Y, So MK, Loening AM, Yao H, Gambhir SS, Rao J. Halo Tag protein-mediated site-specific conjugation of bioluminescent proteins to quantum dots Angew Chem Int Ed Engl 2006; 45(30 ): 4936-0., 56 So MK, Yao H, Rao J. HaloTag protein-mediated specific labeling of living cells with quantum dots Biochem Biophys Res Commun 2008; 374(3 ): 419-23.]. Despite its utility in these applications, when HT2 was fused to more difficult fusion partners the fusions were frequently insoluble or produced at very low levels. Tags including GST and MBP are thought to improve the expression of proteins by promoting the rapid adoption of stable conformations either during or shortly after translation [57 Baneyx F. Recombinant protein expression in Escherichia coli Curr Opin Biotechnol 1999; 10(5 ): 411-21.]. We predicted HT2 was limiting in this regard because of an inability to fold into a thermodynamically stable end product, and used further molecular evolution to optimize HT2 for improved stability. These efforts ultimately produced a variant (HT7) containing 25 amino acid substitutions. In general the individual substitutions provided modest benefits to functional expression, presumably through subtle structural change to the protein. However, the cumulative impact of combining the changes resulted in more significant improvements, consistent with previous reports on the additivity of mutations [58 Wells JA. Additivity of mutational effects in proteins Biochemistry 1990; 29(37 ): 8509-17.-60 Gregoret LM, Sauer RT. Additivity of mutant effects assessed by binomial mutagenesis Proc Natl Acad Sci USA 1993; 90(9 ): 4246-50.]. Although we did not investigate each individual substitution in detail, our structure models suggested three of the more significant changes (A224E, N227D, and K195N) were located on the surface of the protein where they appeared to disrupt positively charged patches. Modifying the charge distribution on the surface of the tag to become more uniformly negative could reduce electrostatic attraction between individual HT7 molecules and reduce the tendency for aggregation [61 Calloni G, Zoffoli S, Stefani M, Dobson CM, Chiti F. Investigating the effects of mutations on protein aggregation in the cell J Biol Chem 2005; 280(11 ): 10607-3., 62 Lawrence MS, Phillips KJ, Liu DR. Supercharging proteins can impart unusual resilience J Am Chem Soc 2007; 129(33 ): 10110-2.]. The modification to the tag’s C-terminus (Pro-Ala-Leu-C to Ser-Thr-Leu-Glu-Ile-Ser-Gly-C) likely had a significant impact on the ability of the tag to be fused to the N-terminus of a partner protein. The additional Glu (position 294) may function to stabilize the α-helix in this region by providing hydrogen bonding to adjacent secondary structure elements in the tag. In the absence of any appendage, the tag is unable to form such interactions. LinB, the dehalogenase from Sphingomonas, contains an Arg at the equivalent of position 294 in HT7, and its crystal structure [63 Marek J, Vevodova J, Smatanova IK, Nagata Y, Svensson LA, Newman J, et al. Crystal structure of the haloalkane dehalogenase from Sphingomonas paucimobilis UT26 Biochemistry 2000; 39(46 ): 14082-6.] indicates it forms a hydrogen bond network with four residues from three different adjacent secondary structure elements, effectively tying the C-terminus to the remainder of the protein. Although our results point to improved stability and reduced tendency to aggregate as being responsible for the increased expression of HT7 fusions, there are other possible contributing factors. For example, the mutations when combined could result in a more stable mRNA structure, or perhaps more efficient codon usage or the removal of problematic codon pairs [64 Grantham R, Gautier C, Gouy M, Mercier R, Pave A. Codon catalog usage and the genome hypothesis Nucleic Acids Res 1980; 8(1 ): r49-62.-66 Makrides SC. Strategies for achieving high-level expression of genes in Escherichia coli Microbiol Rev 1996; 60(3 ): 512-38.]. Two of the mutations (A172T, A167V) clearly provided further improved ligand binding kinetics. Our structure models indicate Ala172 is within 3 Å of bound ligand, suggesting a change to Thr may facilitate ligand entry by introducing favorable hydrogen bonding interactions with the ether oxygens of the ligand.
Our final step in the optimization process was to engineer a customized linker sequence that would help spatially separate HT7 from its fusion partner. We were also hopeful that a linker sequence could be identified that would provide structural stabilization to HT7 fusions, protect full-length HT7 fusions from non-specific proteolytic degradation, and promote efficient cleavage by TEV protease for applications where it would be desirable to remove the tag. We incorporated components of the native TEV sequence, previously identified TEV site mutations and some random sequence to the linker and screened for the desired properties. The linkers identified as being best for each orientation (N or C-terminal tag) provided reduced degradation, better TEV cleavage, and the additional benefit of further improved expression for some fusions. The amino acid composition of the best linkers suggests that some degree of structure in this region may be preferred for optimal performance compared to the flexible linkers (Ser/Gly-containing) used in our original constructs.
In summary, HT7 (referred to generally as HaloTag) and its binding ligand represent a novel protein tag system engineered to possess features critical to the optimal performance of a fusion tag for a variety of applications. Unlike other tags HT7 was engineered to have specific design features: structural compatibility as a fusion partner, and the ability to form an essentially irreversible attachment to its modular binding ligand. These features ultimately provide more efficient protein labeling and capture compared to equilibrium based affinity tags [42 Ohana RF, Encell LP, Zhao K, et al. HaloTag7: A genetically engineered tag that enhances bacterial expression of soluble proteins and improves protein purification Protein Expression Purif 2009; 68(1 ): 110-20., 43 Ohana RF, Hurst R, Vidugiriene J, Slater MR, Wood KV, Urh M. HaloTag-based purification of functional human kinases from mammalian cells Protein Expression Purif 2011; 76(2 ): 154-64.]. The utility of HT7 as a “handle” for protein pull-downs was clearly evident by the isolation and functional analysis of one of the largest macromolecular structures, the ribosome. HT7 has also been used with success to coat glass slides to create protein arrays [67 Hurst R, Hook B, Slater MR, Hartnett J, Storts DR, Nath N. Protein- protein interaction studies on protein arrays: effect of detection strategies on signal-to-background ratios Anal Biochem 2009; 392(1 ): 45-53.]. In addition to applications involving protein purification or pull-downs, HT7 has been used as an effective tool for the optical imaging of cells. For example, it has been utilized to achieve spatiotemporal resolution in chromophore-assisted light inactivation (CALI) [68 Takemoto K, Matsuda T, McDougall M, et al. Chromophore-assisted light inactivation of HaloTag fusion proteins labeled with eosin in living cells ACS Chem Biol 2011; 6(5 ): 401-6.], for super-resolution imaging using photoactivatable fluorophores [38 Lee H-lD, Lord SJ, Iwanaga S, Zhan K, Xie H, Williams JC, et al. Superresolution imaging of targeted proteins in fixed and living cells using photoactivatable organic fluorophores J Am Chem Soc 2010; 132(43 ): 15099-01.], as a probe for magnetic resonance imaging [69 Strauch RC, Mastarone DJ, Sukerkar PA, Song Y, Ipsaro JJ, Meade TJ. Reporter protein-targeted probes for magnetic resonance imaging J Am Chem Soc 2011; 133(41 ): 16346-9.], and for positron emission tomography (PET) [70 Hong H, Benink HA, Zhang Y, et al. HaloTag: a novel reporter gene for positron emission tomography Am J Transl Res 2011; 3(4 ): 392-403.]. HT7 shows comparable versatility to other protein tags in terms of the type of functionality, whether it be a chemical probe or a solid support, that can be attached to a protein [71 Marks KM, Nolan GP. Chemical labeling strategies for cell biology Nat Methods 2006; 3(8 ): 591-6.-75 Gallagher SS, Sable JE, Sheetz MP, Cornish VW. An in vivo covalent TMP-tag based on proximity-induced reactivity ACS Chem Biol 2009; 4(7 ): 547-6.]. However, in contrast to most other tags HT7 offers the ability to bind customized ligands containing user-defined functionalities, which enables its utility as a single genetic construct that can be used for a variety of in vitro and in vivo applications. Since its development and commercialization HT7 has become a valuable research tool for a broad range of applications including imaging, protein purification, and the study of protein interactions.
SUPPLEMENTARY MATERIAL
Supplementary Material is available on the publisher’s website along with the published article.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
We are grateful to Dan Simpson for performing gel permeation chromatography, Sergei Saveliev and Nidhi Nath for insightful discussions, and Michele Arduengo and Mary Hall for critical reading of the manuscript. We also thank Gregg Colwell at Gene Dynamics, LLC for help with vector constructions, NextGen Sciences and Grzegorz Sabat at the University of Wisconsin-Madison for performing mass spectrometry analysis, and Darrell McCaslin at the University of Wisconsin-Madison for performing CD analysis.