- Home
- About Journals
-
Information for Authors/ReviewersEditorial Policies
Publication Fee
Publication Cycle - Process Flowchart
Online Manuscript Submission and Tracking System
Publishing Ethics and Rectitude
Authorship
Author Benefits
Reviewer Guidelines
Guest Editor Guidelines
Peer Review Workflow
Quick Track Option
Copyediting Services
Bentham Open Membership
Bentham Open Advisory Board
Archiving Policies
Fabricating and Stating False Information
Post Publication Discussions and Corrections
Editorial Management
Advertise With Us
Funding Agencies
Rate List
Kudos
General FAQs
Special Fee Waivers and Discounts
- Contact
- Help
- About Us
- Search


The Open Leukemia Journal
(Discontinued)
ISSN: 1876-8164 ― Volume 5, 2013
Induction of Apoptosis in Human Leukemic Cell Lines U937, K562 and HL-60 by Litchi chinensis Leaf Extract Via Activation of Mitochondria Mediated Caspase Cascades
Soma Royaa, Shila E. Besraa, Tripti Deb, Bisweswar Banerjeec, Joydeep Mukherjeed, Joseph R. Vedasiromoni*, a
Abstract
The apoptogenic activity of Litchi chinensis leaf extract (LCLE) was investigated against three human leukemic cell lines-U937, K562 and HL-60. LCLE inhibited cell growth and metabolic activity of the leukemic cells and showed characteristic features of apoptosis. Flow cytometric analysis showed appreciable number of cells in early and late apoptotic stages. While U937 and K562 cell populations were arrested in the G2-M phase, the HL-60 cell population was arrested in the G1 phase of cell cycle. LCLE induced apoptosis is mediated through mitochondrial intrinsic pathway involving the release of cytochrome c into the cytosol and activation of caspase-9 and caspase-3
Article Information
Identifiers and Pagination:
Year: 2008Volume: 1
First Page: 1
Last Page: 14
Publisher Id: TOLEUKEMIAJ-1-1
DOI: 10.2174/1876816400901010001
Article History:
Received Date: 9/10/2008Revision Received Date: 17/10/2008
Acceptance Date: 22/10/2008
Electronic publication date: 14 /11/2008
Collection year: 2008
open-access license: This is an open access article licensed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted, non-commercial use, distribution and reproduction in any medium, provided the work is properly cited.
* Address correspondence to this author at the Drug Development Division, Indian Institute of Chemical Biology, 4, Raja S.C. Mullick Road, Kolkata-700032, India; Fax: 91-33-2473-0284/1597; E-mail: j_rajan_49@yahoo.com, rajan@iicb.res.in
| Open Peer Review Details | |||
|---|---|---|---|
| Manuscript submitted on 9-10-2008 |
Original Manuscript | Induction of Apoptosis in Human Leukemic Cell Lines U937, K562 and HL-60 by Litchi chinensis Leaf Extract Via Activation of Mitochondria Mediated Caspase Cascades | |
1. INTRODUCTION
Drug discovery from medicinal plants has played a very important role in the treatment of cancer [1Barclay AS, Perdue RE Jr. Distribution of anticancer activity in higher plants Cancer Treat Rep 1976; 60: 1081-13.-5Kim J, Park EJ. Cytotoxic anticancer candidates from natural sources Curr Med Chem Anticancer Agents 2002; 2: 485-537.]. Litchi chinensis is a medium-sized evergreen tree, 15-20 m tall, extensively cultivated in the native regions of China and also elsewhere in south-east Asia and more recently in United States, eastern Australia, South Africa and Mexico mainly for its sweet fruits and also grown as an ornamental tree. The seeds of the tree were used as anodyne and prescribed in neuralgic disorders and orchitis [6Kirtikar KR, Basu BD. Indian Medicinal Plants Allahabad (India) Lalit Mohan Basu Publications 1933; 1: 636-7.]. The litchi fruit pericarp extract and its constituents have been reported to possess anti-cancer activity against human breast cancer and hepatocellular carcinoma in vitro and in vivo [7Wang X, Yuan S, Wang J, et al. Anticancer activity of litchi fruit pericarp extract against human breast cancer in vitro and in vivo Toxicol Appl Pharmacol 2006; 215: 168-78., 8Wang X, Wei Y, Yuan S, Liu G, Zhang YL. Wang WPotential anticancer activity of litchi fruit pericarp extract against hepatocellular carcinoma in vitro and in vivo Cancer Lett 2006; 239: 144-50.]. The only study with litchi leaf extract has reported the anti-inflammatory, analgesic and antipyretic activities of the petroleum ether extract [9Besra SE, Sharma RM, Gomes A. Antiinflammatory effect of petroleum ether extract of leaves of Litchi chinensis Gaertn (Sapindaceae) J Ethnopharmacol 1996; 54: 1-6.]. It is well known that tumor promoters recruit inflammatory cells to the application site and cancer development may act by aggravating inflammation in the tissue and vice versa [10Rosin MP, Anwar WA, Ward AJ. Inflammation, chromosomal instability and cancer the schistosomiasis model Cancer Res 1994; 54: 1929S-33S.]. It is also reported that inflammatory cells are capable of inducing genotoxic effects [10Rosin MP, Anwar WA, Ward AJ. Inflammation, chromosomal instability and cancer the schistosomiasis model Cancer Res 1994; 54: 1929S-33S.]. Moreover, some anti-inflammatory chemopreventive agents have been found to suppress growth and proliferation of transformed or malignant cells through induction of programmed cell death or apoptosis [11Smaha HS, Kelloff GJ, Steele V, Rao CV, Reddy BS. Modulation of apoptosis by sulindac. curuminphenylethyl-3-methylcaffeate and 6-phenylhexyl isothiocyanateapoptotic index as a biomarker in colon cancer chemoprevention and promotion Cancer Res 1997; 57: 1301-5., 12Bellosillo B, Pique M, Barragan M, et al. Aspirin and salicylate induce apoptosis and activation of caspases in B-cell chronic lymphocytic leukemia cells Blood 1998; 92: 1406-.]. So it is likely that anti-inflammatory agents may possess anti-cancer activity and vice versa.
Since it has already been reported that Litchi chinensis leaf extract possesses anti-inflammatory activity, it is plausible that it may possess anti-cancer activity as well. The purpose of the present study was to investigate the cytotoxic and apoptogenic activities of the leaf extract of Litchi chinensis in human leukemic cell lines and to make efforts to identify the possible mechanism of action involved in the anti-leukemic activity.
2. MATERIALS AND METHODS
2.1. Chemicals
RPMI 1640 was purchased from Gibco, USA, Caspase-9 assay kit from US Biological, fetal bovine serum, streptomycin penicillin, L-glutamine, HEPES, Trypan blue, MTT 3-(4,5-dimethylthiozol-2-il)-2,5-2,5-dipheniltetrazoliumbromide, cytosine-arabinoside (Ara-C), acridine orange, ethidium bromide, agarose, ethylene diamine tetraacetic acid (EDTA), proteinase K, propidium iodide (PI), Hoescht 33342, annexin-V FITC, RNase A, Caspase-3 assay kit and Cytochrome c oxidase assay kit were purchased from Sigma-Aldrich, USA. All other chemicals and reagents were of analytical grade and purchased locally.
2.2. Cell Cultures
U937 (human leukemic monocyte lymphoma cell line), K562 (human myelogenous leukemia cell line) and HL-60 (human promyelocytic leukemia cell line) were obtained from National Facility for Animal Tissue and Cell Culture, Pune, India. The cells werecultured and routinely maintained in RPMI 1640 medium supplemented with 10% heat inactivated fetal bovine serum and were incubated at 37 °C in a humidified atmosphere containing 5% CO2 inside a CO2 incubator.
2.3. Extraction and Preparation of Test Sample
The leaves of Litchi chinensis were collected by local supplier from sites around Kolkata, India, during June 2006. The plant was identified by Indian Botanical Garden, Howrah, India, where a voucher specimen (SR-002) has been kept. One kg of Litchi chinensis leaves were sun-dried, ground and soaked in 3 l of 50% aqueous methanol for one week at room temperature (28-34ºC) with occasional shaking. The supernatant was filtered and to the residue 1 l of 50% aqueous methanol was added and kept for another one week. The methanol portion in the whole supernatant was evaporated using a rotary evaporator and then it was lyophilized to remove the water. The solid dark brown residue (35 g) thus obtained was designated as ‘Litchi chinensis leaf extract (LCLE)’ and was kept at 4ºC. From 1 mg/ml stock solution of LCLE in phosphate buffered saline (PBS), different suitable concentrations like 50, 100 and 150 μg/ml were used for different in vitro experiments.
2.4. Cell Growth Inhibition Study and Cytotoxicity Study
U937, K562 and HL-60 cells (1x106) were seeded in 96-well sterile plates and were treated with different concentrations (50, 100, 150 μg/ml) of LCLE for 24, 48 and 72 h. The cell growth inhibition studies were done by trypan blue exclusion method [13Sur P, Chatterjee SP, Roy P, Sur B. 5-Nitrofuran derivatives of fatty acid hydrazides induce differentiation in human myeloid leukemic cell lines Cancer Lett 1995; 94: 27-32.] and the cytotoxicity studies were performed by MTT assay [14Cao Z, Li Y. Chemical induction of cellular antioxidants affords marked protection against oxidative injury in vascular smooth muscle cells Biochem Biophys Res Commun 2002; 292: 50-7.].
2.5. Morphological Studies for Detection of Apoptosis
2.5.1. Fluorescence Microscopic Studies
U937, K562 and HL-60 cells (1x106) treated with 100 μg/ml of LCLE for 24 h were observed using a fluorescence microscope for morphological changes. The untreated control cells and the LCLE treated cells were harvested separately, washed with PBS and then stained with acridine orange (100 μg/ml) and ethidium bromide (100 μg/ml) (1:1). The cells were then immediately mounted on slides and observed under a fluorescence microscope for the morphological determination of the cells undergoing apoptosis.
2.5.2. Confocal Microscopic Studies
Leukemic cells (1x106) were treated with 100 μg/ml of LCLE for 24 h. After 24 h the untreated control cells and LCLE treated cells were harvested and washed with ice cold PBS. The cells were then stained with 10 μg/ml of propidium iodide and 10 μg/ml of Hoescht 33342 separately for 5 min. After mounting on slides the cells were observed to see the differences in nuclear morphology between the untreated and the LCLE treated leukemic cells under confocal laser scanning microscope (Leica TCS-SP2 system, Leica Microsystem, Heidelberg, Germany) installed with an inverted microscope [Leica DM-7RB] as per Mishell et al’s method [15Mishell BB, Shiigi SM, Henry C, Eds. Preparation of mouse cell Suspensions.In: Mishel BB.Shiigi SM., Selected Methods in Cellular Immunology 23 San Francisco.: Freeman WH & Co 1980; p. 21.]. Images for propidium iodide and Hoescht 33342 were acquired from argon/krypton laser and UV laser line using 590 nm long pass filter for propidium iodide and 450 nm band pass filter for UV images.
2.5.3. Study of Phosphatidylserine (PS) Externalization
PS externalization was examined after treating the cells (1x106) with 100 μg/ml of LCLE for 24 h under confocal laser scanning microscope (Leica TCS-SP2 system, Leica Microsystem, Heidelberg, Germany). The untreated and the LCLE treated cells were harvested separately, washed with ice cold PBS and annexin V FITC binding buffer (10 mM HEPES, 140 mM NaCl and 2.5 mM CaCl2 2H2O; pH 7.4) respectively and they were then stained with 5 μl of annexin V FITC for 10 min at room temperature. The cells were mounted on slides and the images were captured to observe the cells undergoing early apoptosis.
2.6. Detection of Apoptosis by DNA Fragmentation and Agarose Gel Electrophoresis
U937, K562 and HL-60 cells were treated with 100 μg/ml of LCLE and with standard anti-cancer drug AraC (200 μg/ml) for 24 h. The cells were harvested and washed twice with PBS. The cells were resuspended in 500 μl of lysis buffer (50 mM Tris-HCl, pH -8.0, 10 mM EDTA, 0.5% SDS), 100 μg/ml of proteinase K was added and incubation was done at 50 °C for 1 h and 37 °C overnight respectively. DNA extraction was done by following the general phenol-chloroform extraction procedure [16Herrmann H, Lorenz HM, Voll R, Grunke M, Woith W, Kalden JR. A rapid and simple method for the isolation of apoptotic DNA fragments Nucleic Acids Res 1994; 22: 5506-7.] and kept at -20 °C overnight. After centrifugation, DNA precipitates were washed with 70% ethanol, dried and evaporated at room temperature and dissolved in TE buffer (pH 8.0) at 4 °C overnight. To detect the DNA fragments, the isolated DNA samples were electrophoresed overnight at 20 V in 1% agarose gel and stained with ethidium bromide. DNA fragmentation was observed in UV transilluminator.
2.7. Detection of Apoptosis by Flow Cytometric Analysis
In order to investigate the type of cell death induced by LCLE, flow cytometric analysis was done by performing dot plot assay. The leukemic cells (2x106) were treated with 150 μg/ml of LCLE for 24 h. The cells were pelleted down, centrifuged at 2000 rpm for 8 min at 4 °C and washed with annexin V FITC binding buffer (10 mM HEPES, 140 mM NaCl and 2.5 mM CaCl2 2H2O; pH 7.4). Again after centrifuging at 2000 rpm at 4 °C, the cell pellets were dissolved in annexin V FITC binding buffer containing annexin V FITC and propidium iodide. After 15 min incubation in dark at room temperature flow cytometric analysis was done. All data were acquired with a Becton-Dickinson FACS Caliber single laser cytometer. Flow-cytometric reading was taken using 488 nm excitation and band pass filters of 530/30 nm (for FITC detection) and 585/42 nm (for PI detection). Live statistics were used to align the X and Y mean values of the Annexin-V FITC or PI stained quadrant populations by compensation. Data analysis was performed with Cell Quest (Macintosh platform) program.
2.8. Study of Cell Cycle Arrest by Flow Cytometric Analysis
2x106 cells were treated with 150 μg/ml of LCLE for 48 h. Cells were washed with PBS, fixed with cold methanol by adding methanol drop-wise and kept at -20 °C for 3 min. They were then resuspended in cold PBS and kept at 4 °C for 90 min. Cells were pelleted down, dissolved in cold PBS, treated with RNase A for 30 min at 37 °C and stained with propidium iodide (20 μl from 50 μg/ml) and kept in dark for 15 min. Cell cycle phase distribution of nuclear DNA was determined on FACS, fluorescence detector equipped with 488 nm argon laser light source and 623 nm band pass filter (linear scale) using Cell Quest software (Becton Dickinson). Analysis of flowcytometric data was performed using ModFit software.
2.9. Cytochrome c Oxidase Assay
U937, K562 and HL-60 cells (1x106) were treated with LCLE (50, 100 and 150 µg/ml) for 24 h. The cells were harvested, lysed and then cytosolic extracts were prepared separately for untreated control and LCLE treated U937, K562 and HL-60 cells. The cytochrome c oxidase assay was performed according to the instructions provided in the kit. 20 µl of cytosolic extract was added to 950 µl of 1X assay buffer (10 mM Tris-HCl, pH 7.0, containing 120 mM KCl). The reaction volume was made to 1.05 ml with 1X enzyme dilution buffer (10 mM Tris-HCl, pH 7.0, containing 250 mM sucrose) and was mixed by inversion. The reaction was started by the addition of 50 µl of Ferrocytochrome c substrate solution and mixed by inversion. The O.D was read immediately at 550 nm.
2.10. Caspase-9 Assay
The assay was performed using a Caspase-9, Apoptosis Detection, Colorimetric BioAssay Kit (US Biological) according to the manufacturer’s protocol. U937, K562 and HL-60 cells (1x106) were treated with 50, 100 and 150 µg/ml of LCLE for 24 h. The cells were pelleted down and resuspended in 50 µl of cell lysis buffer (supplied with the kit) and incubated on ice for 10 min. After centrifuging at 10,000 x g for one min, the supernatants (cytosolic extract) were transferred to fresh tubes and kept on ice and the caspase-9 assay was performed according to the supplied kit protocol. 50 µl of 2X reaction buffer (containing 10 mM DTT) was added to each sample. 5 µl of LEHD-pNA substrate (4 mM) (200 µM final concentration) was added and incubation was done at 37 °C for 1-2 h. Absorbance was read at 405 nm and calculations were thereby done.
2.11. Caspase-3 Assay
The assay was performed using a Caspase-3 Assay kit, Colorimetric (Sigma) according to the manufacturer’s protocol. U937, K562 and HL-60 cells (1x106) were treated with 50, 100 and 150 μg/ml of LCLE for 24 h. The untreated control and the treated cells were pelleted down by centrifugation at 600 x g for 5 min at 4 °C. Supernatants were removed and the cell pellets were washed with 1 ml of PBS. The cells were again centrifuged and the supernatants were removed completely. The cell pellets were suspended in 100 µl of 1X lysis buffer (50 mM HEPES, pH 7.4, 5 mM CHAPS, 5 mM DTT) and incubated on ice for 20 min. The lysed cells were centrifuged at 20,000 x g for 15 min at 4 °C and the supernatants (cell lysates) were analysed for the caspases-3 activity according to the manufacturer’s protocol. Cell lysates were incubated with 2 mM Caspase-3 substrate (Ac-DEVD-pNA) in 1X assay buffer (20 mM HEPES, pH 7.4, 2 mM EDTA, 0.1% CHAPS, 5 mM DTT) for 90 min at 37 °C. The absorbance was read at 405 nm and the results were calculated using a p-nitroaniline calibration curve.
2.12. Statistical Analysis
All data are represented as arithmetic mean ± S.E.M. Statistical analysis was done by Student’s t-test. A probability value of less than 0.05 was chosen as the criterion of statistical significance.
3. RESULTS
3.1. Cell Growth Inhibition Study and Cytotoxicity Study
LCLE at concentrations of 50, 100, 150 μg/ml significantly inhibited the growth of U937, K562 and HL-60 cells compared with that of the control cells after 24, 48 and 72 h of treatment in a concentration-dependent manner (Fig. 1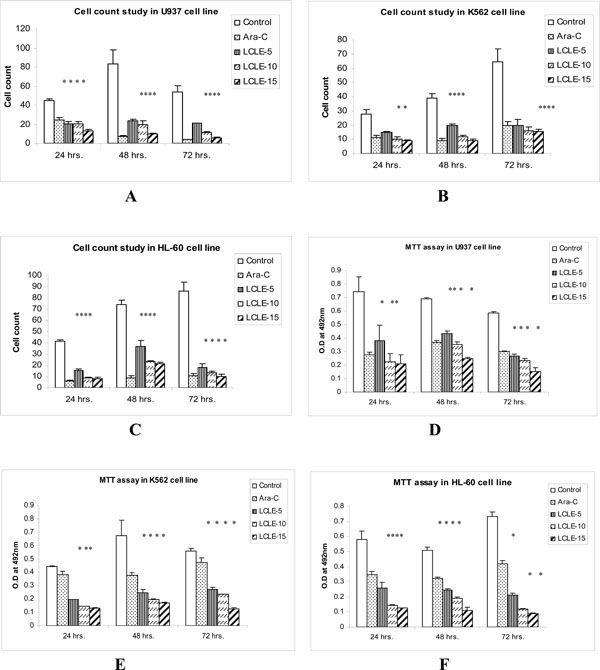 ). In the MTT assay, there was significant concentration-dependent reduction in the O.D values after treating the U937, K562 and HL-60 cells with 50, 100, 150 µg/ml of LCLE for 24, 48 and 72 h compared to the control cells (Fig. 1). These observations provided proof for cytotoxic nature of LCLE.
). In the MTT assay, there was significant concentration-dependent reduction in the O.D values after treating the U937, K562 and HL-60 cells with 50, 100, 150 µg/ml of LCLE for 24, 48 and 72 h compared to the control cells (Fig. 1). These observations provided proof for cytotoxic nature of LCLE.

3.2. Morphological Studies for Detection of Apoptosis
3.2.1. Fluorescence Microscopic Studies
Observations revealed that LCLE treated leukemic cells (U937, K562 and HL-60) were stained with both acridine orange and ethidium bromide compared with that of the untreated control cells, stained with only acridine orange, indicating the fact that the treatment with LCLE brought about apoptotic changes in the cells like condensation of chromatin and nuclear fragmentation (Fig. 2 ).
).

3.2.2. Confocal Microscopic Studies
LCLE induced apoptotic changes in all the three leukemic cells after 24 h of treatment showing chromatin disintegration and formation of apoptotic bodies whereas the untreated control cells were with intact nuclei (Fig. 3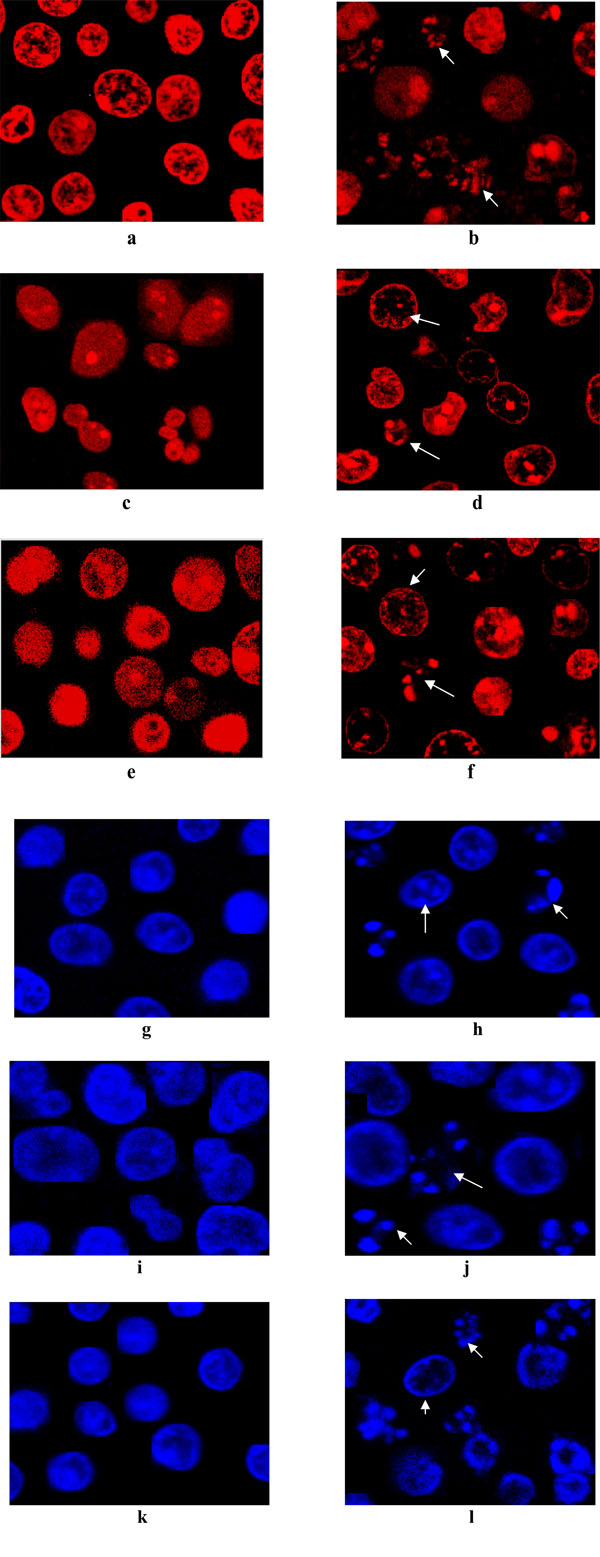 ).
).

3.2.3. Study of Phosphatidylserine (PS) Externalization
Phosphatidylserine is predominantly accumulated in the inner leaflet of plasma membrane of living cells but in the apoptotic cells, phosphatidylserine is translocated from inner to outer leaflet of plasma membrane. Treatment with LCLE caused externalization of phosphatidylserine which after binding with annexin V FITC gave green fluorescence (Fig. 4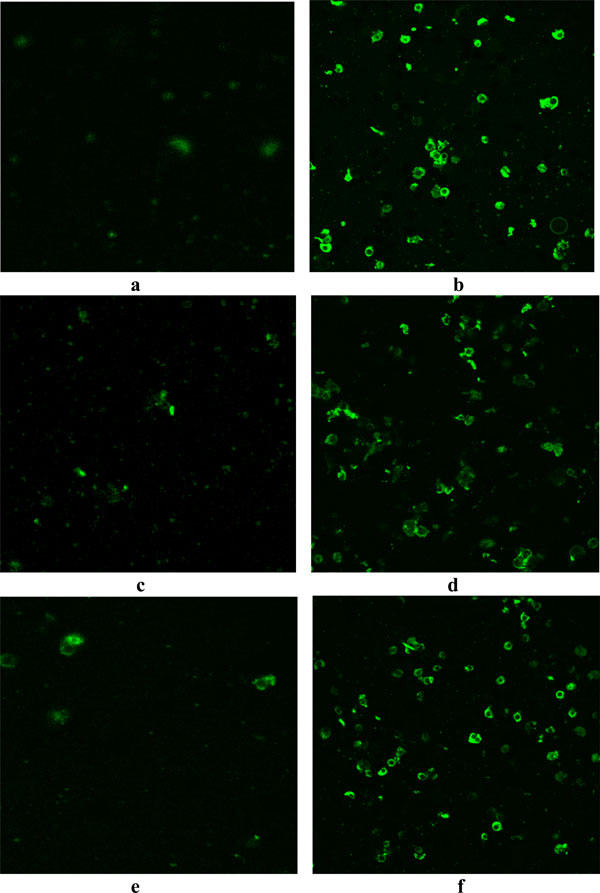 ).
).

3.3. Detection of Apoptosis by DNA Fragmentation and Agarose Gel Electrophoresis
The gel pattern of the DNA samples isolated from untreated control U937, K562 and HL-60 cells showed intact DNA bands whereas the gel pattern of the DNA samples isolated from LCLE treated U937, K562 and HL-60 cells showed degraded DNA bands in the form of ladders (Fig. 5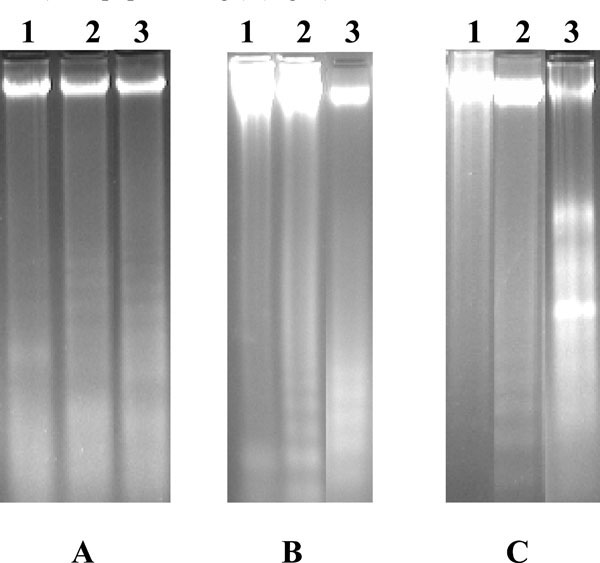 ). So, the observations confirmed that the treatment with LCLE caused apoptosis in all the three human leukemic cells i.e., U937, K562 and HL-60.
). So, the observations confirmed that the treatment with LCLE caused apoptosis in all the three human leukemic cells i.e., U937, K562 and HL-60.

3.4. Detection of Apoptosis by Flow Cytometric Analysis
In the flow cytometric analysis, double labeling technique, using annexin V FITC and propidium iodide, was utilized. Lower left (LL) quadrant (annexin V-/PI-) is regarded as the population of live cells, lower right quadrant (LR) (annexin V+/PI-) is considered as the cell population at early apoptotic stage, upper right (UR) quadrant (annexin V+/PI+) represents the cell population at late apoptotic stage and upper left (UL) quadrant (annexiv V-/PI+) is considered as necrotic cell population. Flow cytometric data analysis revealed that after 24 h of treatment with LCLE, 12.11% of U937, 23.16% of K562 and 9.21% of HL-60 cells were in LR quadrant (early apoptotic stage) and 4.64% of U937 and 27.65% of K562 and 24.42% HL-60 cells were in UR quadrant (late apoptotic stage) (Fig. 6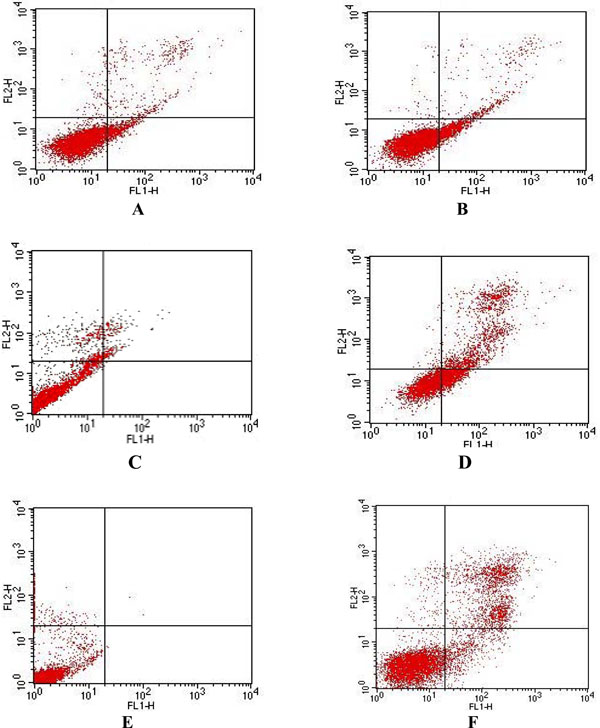 ).
).

3.5. Study of Cell Cycle Arrest by Flow Cytometric Analysis
The flow cytometric analysis showed that when compared with that of the untreated control U937 and K562 cells, 48 h of LCLE treatment caused a progressive increase in the number of cells with a 4N and with intermediate DNA contents i.e. cells with an irregular DNA distribution, characteristic of apoptotic cell populations. These observations revealed that LCLE treatment inhibited the growth of U937 and K562 cells by arresting the cell populations in the G2-M phase of the cell cycle (Fig. 1). But in case of HL-60 cells, after 48 h of LCLE treatment, there was a sharp increase in the number of cells with a 2N DNA content when compared with that of the untreated control cells, indicating that the LCLE treatment inhibited the growth of HL-60 cells by cell cycle arrest in the G1 phase (Fig. 7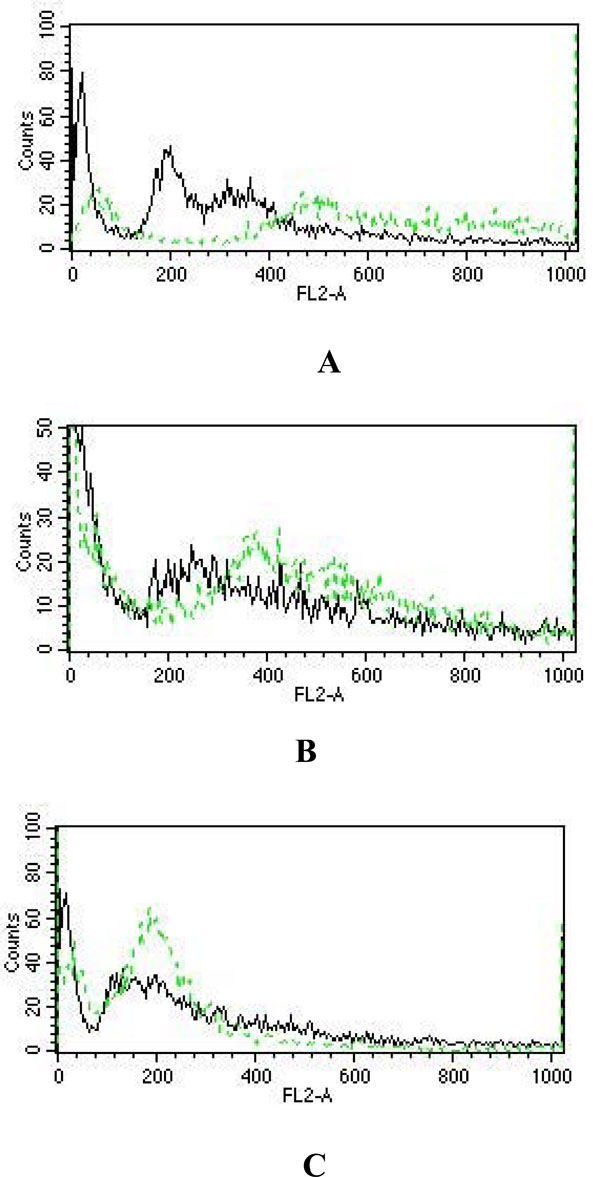 ).
).

3.6. Cytochrome c Oxidase Assay
The cytochrome c oxidase colorimetric assay is based on observations of the decrease in absorbance at 550 nm of ferrocytochrome c caused by its oxidation to ferricytochrome c by cytochrome c oxidase. The cytosolic extracts of control untreated U937, K562 and HL-60 cells showed higher O.D values compared with that of LCLE treated cells indicating decreased oxidation of ferrocytochrome c to ferricytochrome c or in other words, lesser activity of cytochrome c oxidase whereas the decreased O.D values of the cytosolic extracts of LCLE treated U937, K562 and HL-60 cells implied greater cytochrome c oxidase activity resulting in the increased oxidation of ferrocytochrome c to ferricytochrome c (Fig. 8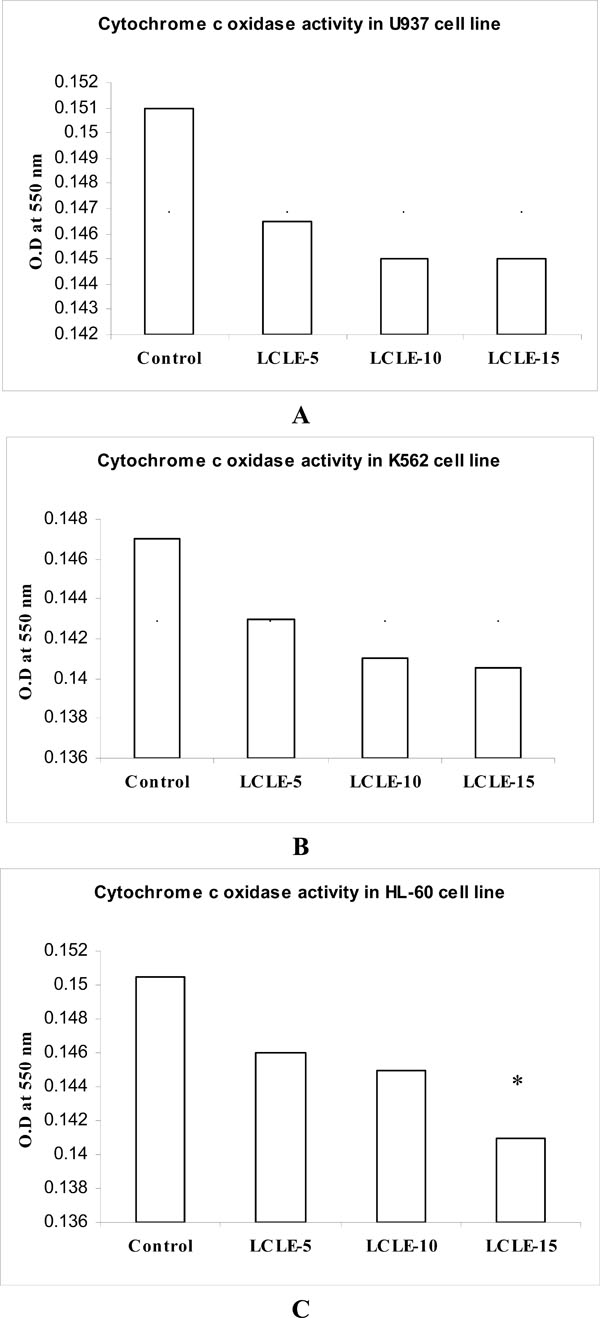 ). Treatment with 50, 100 and 150 µg/ml of LCLE showed concentration-dependent response in all the three leukemic cell lines (Fig. 8). It might be plausible that increased cytochrome c oxidase activity in the cytosolic extracts of LCLE treated U937, K562 and HL-60 cells was associated with the process of cell death.
). Treatment with 50, 100 and 150 µg/ml of LCLE showed concentration-dependent response in all the three leukemic cell lines (Fig. 8). It might be plausible that increased cytochrome c oxidase activity in the cytosolic extracts of LCLE treated U937, K562 and HL-60 cells was associated with the process of cell death.

3.7. Caspase-9 Assay
Caspase-9 is one of the main initiator caspases and has been linked to the mitochondrial death pathway. To investigate whether the treatment with LCLE induced apoptosis via intrinsic pathway, caspase-9 assay was performed in U937, K562 and HL-60 cells. The experiments revealed concentration-dependent increase in the caspase-9 activity in the cytosolic extract of LCLE (50, 100 and 150 µg/ml) treated U937, K562 and HL-60 cells compared with that of the untreated control U937, K562 and HL-60 cells respectively (Fig. 9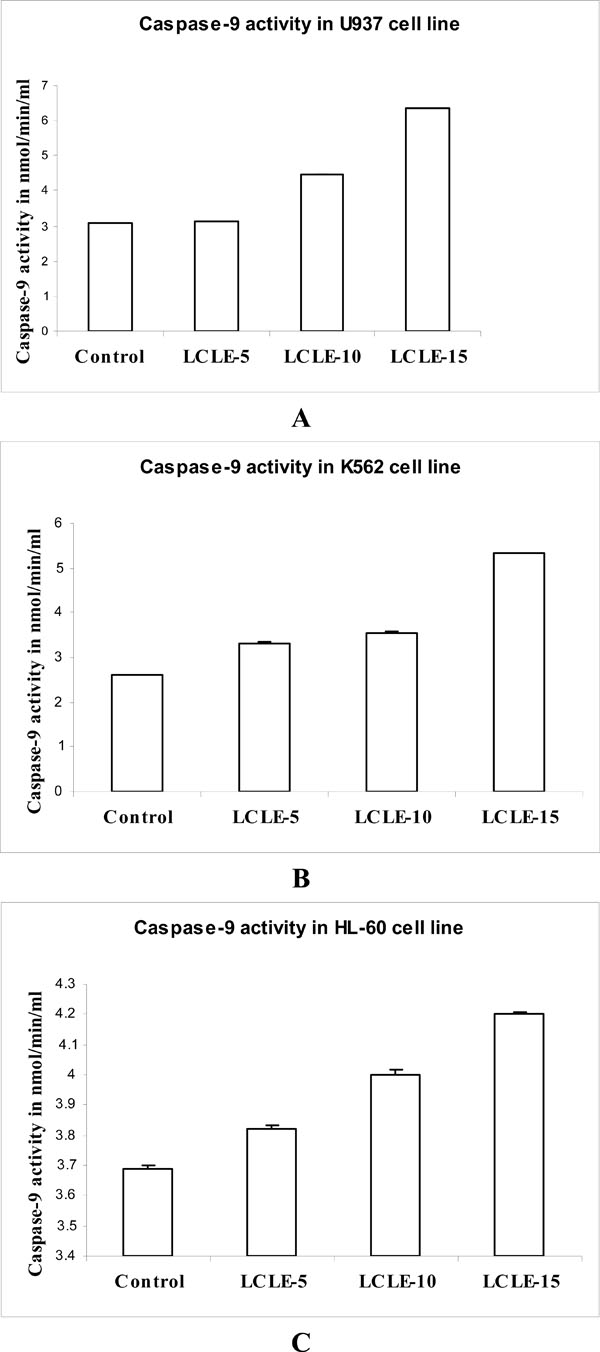 ), supporting the fact that apoptosis induced by the LCLE treatment might be mediated through the intrinsic pathway.
), supporting the fact that apoptosis induced by the LCLE treatment might be mediated through the intrinsic pathway.

3.8. Caspase-3 Assay
Sequential activation of caspases plays a central role in the execution-phase of cell apoptosis. The primary target of the caspase-9 is procaspase-3, one of the most deleterious effector caspases. To observe whether caspase-9 activated caspase-3 after LCLE (50, 100 and 150 µg/ml) treatment, caspase-3 assay was performed in U937, K562 and HL-60 cells. Caspase-3 activation was clearly observed in LCLE treated U937, K562 and HL-60 cells in a concentration-dependent fashion when compared with that of the untreated control cells (Fig. 10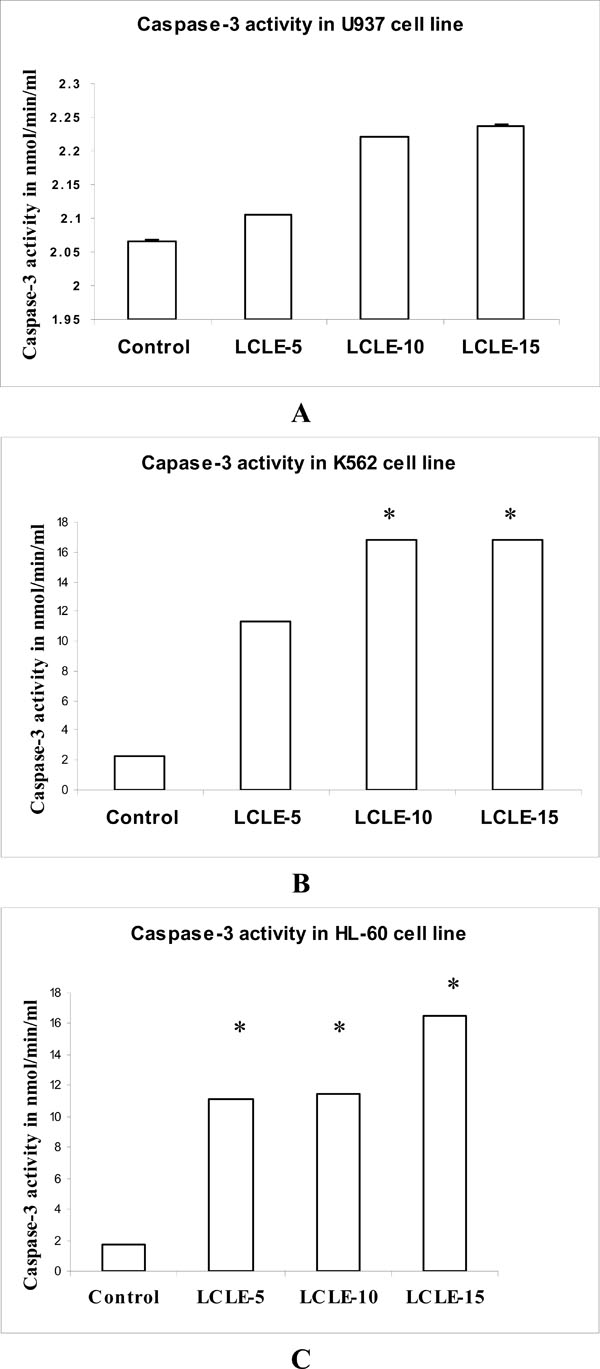 ).
).

4. DISCUSSION
A significant part of drug discovery in the last forty years has been focused on agents to prevent or treat cancer. This is not surprising because, in most developed countries and, to an increasing extent, in developing countries, cancer is amongst the three most common causes of death and morbidity. Treatment for cancer involves surgery, radiotherapy and chemotherapy and often a combination of two or all three is employed [17Houghton P, Fang R, Techatanawat I, Steventon G, Hylands PJ, Lee CC. The sulphorhodamine (SRB) assay and other approaches to testing plant extracts and derived compounds for activities related to reputed anticancer activity Methods 2007; 42: 377-87.]. Natural products provide an appreciable percentage of new active lead molecules, clinical candidates and drugs despite competition from different methods of drug discovery. The number of natural product-derived drugs present in the total drug launches from 1981 to 2002 was recently analysed [18Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981-2002 J Nat Prod 2003; 66: 1022-37.,19Cragg GM, Newman DJ, Snader KM. Natural products in drug discovery and development J Nat Prod 1997; 60: 52-60.] and it was concluded that natural products are still a significant source of new drugs, especially in the anti-cancer and anti-hypertensive therapeutic areas [18Newman DJ, Cragg GM, Snader KM. Natural products as sources of new drugs over the period 1981-2002 J Nat Prod 2003; 66: 1022-37.]. Chemoprevention is a novel approach emphasizing on the prevention or delay of carcinogenesis by means of pharmacological, biological, and nutritional intervention and recently, cancer chemoprevention with strategies using foods and medicinal herbs has been regarded as one of the most visible fields for cancer control [20Jo EH, Hong HD, Ahn NC, et al. Modulations of the Bcl-2/Bax family were involved in the chemopreventive effects of licorice root (Glycyrrhiza uralensis Fisch) in MCF-7 human breast cancer cell J Agric Food Chem 2004; 52: 1715-9.].
Practical experience has shown that once a plant extract is found to possess anti-inflammatory activity, it is better to test whether it possesses any anti-cancer activity because it is now pretty well established that inflammation and cancer go hand in hand. Since the anti-inflammatory activity of Litchi chinensis leaf extract has already been reported [9Besra SE, Sharma RM, Gomes A. Antiinflammatory effect of petroleum ether extract of leaves of Litchi chinensis Gaertn (Sapindaceae) J Ethnopharmacol 1996; 54: 1-6.], it is likely that the extract of Litchi chinensis leaves may also have anti-leukemic activity. The present investigation confirmed the cytotoxic activity and apoptosis inducing ability of aqueous methanolic extract of Litchi chinensis leaves (LCLE) against three human leukemic cell lines- U937, K562 and HL-60. The anti-proliferative and the cytotoxic activities of LCLE were supported by the observations in cell growth inhibition studies and in MTT assays respectively. LCLE inhibited the growth and the metabolic activities of U937, K562 and HL-60 cells in a concentration-dependent manner. In a separate study it was found that LCLE possesses potent immunomodulatory activity and that at concentrations used in the present study it did not show any cytotoxic effect in a macrophage cell line, RAW264.7 as observed in MTT assay. This finding reveals that LCLE preferentially acts on leukemic cells.
Apoptogenic activity of LCLE was investigated by different morphological studies like fluorescence microscopic, confocal microscopic and phosphatidylserine (PS) externalization studies. The process of apotosis is characterized by several morphological changes such as cell shrinkage, membrane blebbing, chromatin condensation, nuclear fragmentation and formation of apoptotic bodies. Fluorescence microscopic images clearly showed nuclear disintegration of LCLE treated leukemic cells compared with that of the untreated control cells when stained with acridine orange and ethidium bromide. The untreated control cells showed bright green fluorescence as the live cells with intact membrane excluded ethidium bromide and only acridine orange could enter into them. On the contrary LCLE treated cells showed more intense orange-red fluorescence and reduced green fluorescence since apoptotic and necrotic cells could not exclude the dyes and gave a combination of orange-red and green fluorescence. So, the observations indicated that the treatment with LCLE was inducing apoptosis in the leukemic cells. Apoptogenic activity of LCLE was further evidenced from the confocal microscopic images of the treated leukemic cells when compared with that of the untreated control cells. After LCLE treatment, U937, K562 and HL-60 cells showed several signs of apoptosis like chromatin condensation, nuclear fragmentation and formation of apoptotic bodies whereas the untreated control cells were with intact nuclei.
Externalization of PS from inner leaflet to outer leaflet of the membrane is the hallmark of early phase of apoptosis. Externally translocated PS binds with annexin V in a calcium dependent manner [21Martin SJ, Reutelingsperger CP, McGahen AJ, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus inhibition by overexpression of Bcl-2 and Abl J Exp Med 1995; 182: 1545-56.]. Fluorescence microscopic images of treated U937, K562 and HL-60 cells showed bright green fluorescent rings of externalized PS, supporting the fact that treatment with LCLE induced apoptosis in the leukemic cells. Further evidence in support of the apoptogenic activity of LCLE was obtained from the gel patterns of agarose gel electrophoresis. LCLE treated cells showed degraded DNA bands in the form of ladders, a typical indication of apoptosis, whereas the untreated control cells showed intact DNA bands when observed in UV transilluminator. Dual staining with annexin V FITC and propidium iodide in dot plot assay made it possible to identify live, early apoptotic and late apoptotic cells [22Darzynkiewicz Z, Bedner E, Smolewski P. Flow cytometry in analysis of cell cycle and apoptosis Semin Hematol 2001; 38: 179-93., 23Wising C, Azem J, Zetterberg M, Svensson LA, Ahlman K, Lagergard T. Induction of apoptosis/necrosis in various human cell lineages by Haemophilus ducreyi cytolethal distending toxin Toxicon 2005; 45: 767-.]. Experiments showed increased number of cells in the early and late apoptotic stage after treatment with LCLE implying the fact that apoptosis was triggered by the treatment with LCLE in U937, K562 and HL-60 cells. Cell cycle analysis revealed that treatment with LCLE arrested the U937 and K562 cell populations in the G2-M phase and HL-60 cell population in the G1 phase of cell cycle.
There are two major apoptotic pathways known to date, initiated by either the mitochondria (the 'intrinsic' pathway) or the cell surface receptors (the 'extrinsic' pathway). Mitochondria-mediated apoptosis occurs in response to a wide range of death stimuli, including activation of tumor suppressor proteins (such as p53) and oncogenes (such as c-Myc), DNA damage, chemotherapeutic agents, serum starvation, and ultraviolet radiation [24Shi Y. A structural view of mitochondria-mediated apoptosis Nat Struct Biol 2001; 8: 394-401.]. In the intrinsic pathway, diverse proapoptotic signals converge at the mitochondrial level, inducing the translocation of cytochrome c into the cytosol. Cytochrome c triggers caspase-9 activation initiating a downstream caspase cascade through the complex formation with Apaf-1, dATP, and pro-caspase-9 in the cytosol, which ultimately lead to the activation of the executioner caspase-3 and finally cell death [25Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis Annu Rev Cell Dev Biol 1999; 15: 269-90.-30Kaufmann SH, Earnshaw WC. Induction of apoptosis by cancer chemotherapy Exp Cell Res 2000; 256: 42-9.].
In the present study, the release of cytochrome c into the cytosol was demonstrated in the cytochrome c oxidase assay. The cytochrome c oxidase activity was increased in a concentration-dependent way after treatment with LCLE (50, 100 and 150 µl) in U937, K562 and HL-60 cells as represented by the decreased O.D values when compared with that of the untreated control cells. Caspase-9 and caspase-3 assays showed concentration-dependent increase in the activities of caspase-9 and caspase-3 respectively after LCLE treatment in U937, K562 and HL-60 cells indicating that the cytochrome c release was a preceding event for the activation of the mitochondria mediated caspase cascades. The involvement of the caspase cascade in the cytotoxic activity of LCLE needs to be confirmed using specific caspase inhibitors.
These observations in the present work suggest that treatment with LCLE induced apoptosis via mitochondria mediated intrinsic pathway in U937, K562 and HL-60 cell populations.
K562 is an erythroblastic cell line expressing the typical hallmark of CML, the Philadelphia (Ph) chromosome, and the B3A2 bcr-abl [31Lozzio CB, Lozzio BB. Human chronic myelogenous leukemia cell line with positive Philadelphia chromosome Blood 1975; 45: 321-4.] and it has been extensively studied in order to investigate CML and the metabolic pathways underlying its therapy. K562 cell line has also been the object of experiments concerning its resistance to apoptosis- inducing drugs [32Martin S, Lennon S, Bonham A, Cotter T. Induction of apoptosis (programmed cell death) in human leukemic cells by inhibitors of RNA protein synthesis J Immunol 1990; 145: 1859-67.]. Since the present work confirmed the apoptogenic activity of LCLE against multi-drug resistant human myelogenous leukemia cell line, K562, it is plausible that it may lead to identification of a novel anti-leukemic agent against CML.
ACKNOWLEDGEMENT
The work was funded by University Grants Commission, India under Minor Research Project Scheme.